 0373-5939925
0373-5939925 2851259250@qq.com
2851259250@qq.com

介尺度视角下的电催化:从界面、隔膜到多孔电极
电化学催化过程主要是由发生在电极界面的液相传质、物种吸附脱附、电子转移以及表面转化等单元步骤串联组成,该过程决定了电化学催化是具有时空多尺度的复杂系统[1-5]。主要表现在量子尺度的电子转移;原子尺度的活性位;分子水平的电化学催化机理;纳米尺度下催化剂与质子导体,微米尺度的催化层,数十微米尺度的扩散层,以及厘米到分米尺度下的多孔电极等。各尺度下的材料结构、反应传递响应时间能否有效匹配、耦合决定了电化学催化过程的效率。据此,理解上述不同结构、层次之间的介尺度行为,以及从介尺度的视角去探寻多级孔电极的构筑、电化学催化机理以及电催化剂和隔膜设计制备等是精确提升电催化性能的关键[6-9]。
以多孔电极的电催化过程为例(图1),提升其电催化行为,必须合理设计原子尺度的活性位,提升其本征活性的同时,提高活性位的利用率[10]。与此同时还需配合优化三相界面;调控增强气液传质的孔道和电子、质子传输通道;最为重要的是构筑可连接微孔与大孔的介孔通道。该介孔通道的构筑不仅保证了反应物与产物的有效扩散和传递,还确保在微孔内聚集的活性位的利用率,从而将具有不同反应速率和响应时间的气液传质与表界面的电化学反应有机耦合,有效提升电化学催化性能。据此,本文将从介尺度的视角介绍多孔电极结构、催化剂表界面活性位构筑与调控、催化剂介尺度结构可控制备策略以及隔膜的介尺度结构调控。
图1

图1 电催化结构中的介尺度特征[10]
Fig.1 The mesoscale characteristics of electrocatalytic structure[10]
1 多级孔互穿网络型多孔电极
重庆大学魏子栋教授和大连化学物理研究所邵志刚研究员、孙公权研究员合作提出了“一个反应、两种导体、三相界面、四种通道”的典型“一、二、三、四”特征,是对燃料电池中气体多孔电极的一个清晰描述(图2)。即阴极发生氧还原反应时,催化层的三相界面(气相O2,液相H2O,固相催化剂)上必须同时具备电子传输通道(导电通道),质子传输通道,以及有序连通各活性位(微孔和小介孔)的反应物气体传输通道和反应产生水的排出通道(大孔和大介孔)。这就需要多孔电极必须具有特定三维几何结构形貌,有序分布各功能化孔道,使催化活性位得以充分利用[11]。高效的气体多孔电极,就是四条通道必须交汇到所有的催化剂颗粒上。然而,对于目前应用较多的碳基商业催化剂来说,其本征活性到构筑的多孔电极活性间的差异还比较大。进一步优化碳基催化剂多孔电极的孔道结构,增加活性位密度同时,提高活性位的利用率以及优化物种的扩散与传质是目前急需解决的。本文针对气体多孔电极催化与物质传递双功能的精确调控,总结了一些高效的策略。
图2

图2 气体多孔电极“一、二、三、四”特征描述示意图
Fig.2 Schematic diagram of gas porous electrode “one, two, three, four” feature
1.1 应力诱导定向收缩制备氮碳多级孔结构
中空材料作为一类特殊的纳米结构材料,优势在于壳层包含丰富传质通道,使得暴露于内表面的活性位点可以得到充分利用[12-14]。Ding等[15]报道了一种以Co基金属有机框架材料(MOF) (ZIF-67)为前体,通过两步热解-氧化合成Co基纳米颗粒嵌入空心氮掺杂碳催化剂的方法。其中,超高的比表面积和丰富的多级孔结构增加了活性位点的暴露,加速了电荷转移,同时MOF形成的中空多孔碳壳增强了催化剂的导电性,便于电子传递和质量扩散。所制备得到的催化剂表现出可媲美商业Pt/C的氧还原反应(ORR)活性[图3(a)~(c)]。除了采用MOF材料作为中空结构的骨架外,利用硬模板也能制备出具有高活性的多孔中空催化剂。Guo等[16]采用SiO2包覆的碳球作为硬模板,结合浸渍和高温热解,将Cu3P和MoP锚定在空心多孔碳载体中制备得到电催化剂(Cu3P/MoP@C)。其在碱性介质中表现出高的电催化活性和卓越的耐久性。大的介孔结构提供了较大的比表面积,有利于内表面的活性位点与电解液接触,加速ORR动力学[图3(d)~(f)]。因此通过调控其内部空间和壳层内的孔结构,可以设计出丰富多样的多级孔材料,从而在增加活性位密度的同时有效提高传质。
图3

图3 (a)基于Co - Co3O4嵌入空心氮掺杂碳催化剂的示意图;(b)Co3O4/HNCP-40催化剂的SEM, TEM, STEM和EDS mapping图像;(c)ORR极化曲线和5000圈老化前后对比[15];(d)Cu3P/MoP@C催化剂的合成示意图;(e)HR-TEM图像;(f)ORR极化曲线和半波电位以及在0.9 V下的动力学电流密度对比曲线[16]
Fig.3 (a) Illustration of Co-Co3O4-based nanoarchitectures embedded in hollow nitrogen-doped carbon polyhedron; (b) SEM, TEM, STEM, and EDS mappings of Co3O4/HNCP-40; (c) ORR polarization curves and before and after 5000 cycles for the ORR[15]; (d) Schematic illustration of preparation Cu3P/MoP@C; (e) HR-TEM image; (f) ORR polarization and the corresponding half-wave potential (E1/2) and kinetic current density (Jk) at 0.9 V[16]
Wang等[17]首次基于物理应力拉伸效应构建了空心结构。探究ZIF8-多巴胺(PDA)核壳结构材料的微观结构演变过程发现[图4(a)],随焙烧温度升高,材料逐渐从实心结构演变至内部少量空心、具有夹层的核壳结构,进一步升高温度,最终成为了十二面体的空心结构(NC15-900℃)。相比之下,当无PDA涂敷层时,纯相ZIF8纳米晶体在焙烧过程中逐渐降解并自发向内收缩得到了具有十二面体的实心颗粒(NCZIF8-900℃)。由于菱形十二面体具有整体对称结构,不同方向收缩均匀,材料整体受力平衡,因此得到的样品能够保持原有形貌。ORR测试表明[图4(b)],具有中空结构的催化剂活性明显优于实心颗粒催化剂。在碱性环境中,具有中空结构的催化剂半波电位达到0.848 V,动力学交换电流密度达到20.3 mA/cm2@0.8 V,并且优于商业Pt/C催化剂。比较BET比表面积发现[图4(c)],虽然NCZIF8-900℃催化剂具有最大的BET比表面积,但是实心的固体结构在很大程度上限制了活性位点的传质和暴露。这些结果充分证明了中空结构中壳层产生的微、介孔有助于充分暴露活性位点,内部核消失产生的大孔可以极大提升传质效率,增加催化剂的ORR性能。
图4
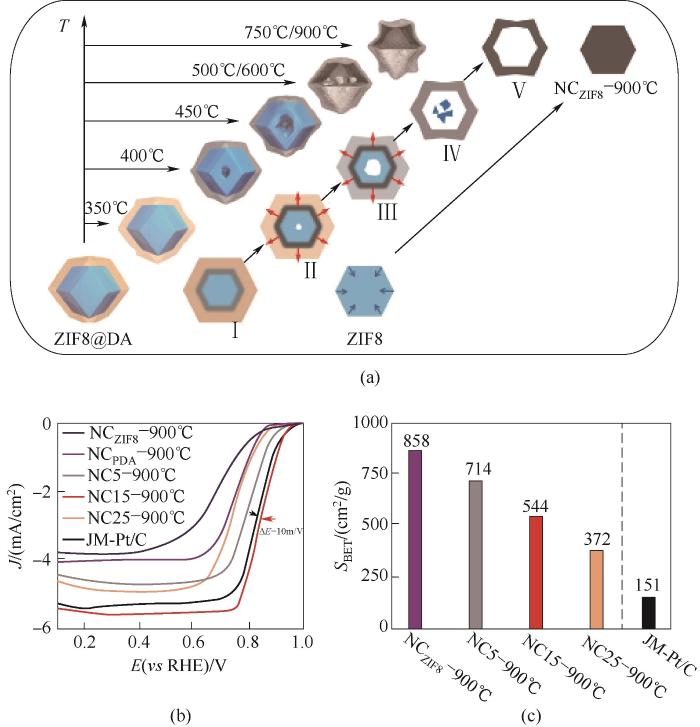
图4 (a)构造空心结构的应力诱导定向收缩机制示意图;(b)在O2饱和的0.1 mol/L KOH 溶液中的ORR极化曲线;(c)催化剂的BET比表面积对比[17]
Fig.4 (a) Schematic illustration of the stresses induced orientation contraction mechanism for constructing the hollow structures; (b) ORR polarization curves measured in O2-saturated 0.1 mol/L KOH solution; (c) The comparison of BET specific surfaces areas[17]
1.2 多级孔互穿网络型多孔电极
催化剂层作为燃料电池的核心,当其发生水淹时,多孔通道中积累的水会直接导致O2传质通道受到压缩,O2分子无法及时到达活性位点,氧还原反应变慢甚至终止[18-21]。通过调控催化剂的孔结构,提升孔体积,能够有效缓解水淹问题。Wang等[22]基于软硬模板法制备了有序大孔-介孔互穿网络抗水淹气体多孔电极,大孔(500 nm)和介孔(13 nm)均呈现规则有序的排布,同时Pt纳米粒子均匀分布在多孔碳载体上,尺寸约为3 nm[图5(a)]。N2吸脱附测试表明多孔电极的孔体积和比表面积分别是商业Pt/C催化剂的3.4倍和4.5倍[图5(b)]。为了研究真实的传质和抗水淹性能,采用“拨浪鼓”结构工作电极对催化剂的传质能力进行评估。如图5(d)所示,“拨浪鼓”电极由一个负载催化剂的Teflon头和一根连接导线组成。测试过程中,Teflon头全部浸入电解质溶液中,同时通入的气体O2扩散至催化剂表面进行反应,产生的电流由金环通过铜线导出。该体系避免了阳极和膜的使用,提供了足够质子,同时厚催化层、气体O2的使用,真实模拟了膜电极(MEA)测试条件。该电极中,影响电流输出的最关键因素即为催化剂层传质效率,通过和某一给定催化剂进行性能比较,即可得到催化剂层传质效率的量化评价。通过对催化剂的传质效率进行定量评价后发现,多孔电极的传质效率是商业Pt/C的4倍[图5(e)]。MEA测试表明,多孔电极的输出功率始终高于商业Pt/C催化剂,即使在强增湿的条件下输出功率也比传统电极提高了45%[图5(c)]。这些结果充分说明孔道调控确实可以有效提高气体多孔电极传质效率与抗水淹能力,加快ORR反应的进行。
图5

图5 (a)催化剂的SEM和TEM图像;(b)催化剂的比表面积和总孔体积比较;(c)MEA性能测试图;(d)自制“拨浪鼓”光学图片和工作示意图;(e)催化剂以10/15 mA/cm2电流密度恒电流放电时的计时电位曲线[22]
Fig.5 (a) SEM and TEM images of the catalysts; (b) Comparison of the specific surface areas and total pore volumes for the two catalysts; (c) Polarization and power densities curves of MEA; (d) The schematic and optical pictures of the self-made rattle-drum -like working electrode; (e) Chronopotentiometry curves recorded at 10/15 mA/cm2, the catalyst was loaded on the “rattle-drum” working electrode [22]
1.3 双孔策略合成高效传质Pt3Co/C氧还原催化剂
在介尺度电催化剂的设计过程中,形成丰富的可达活性位点和快速的传质通道对提高催化活性至关重要。然而,目前应用较多的Pt合金纳米粒子负载在多级孔孔内的方法会导致传质通道变窄、纳米颗粒在孔道内壁的附着力较弱等问题[23-25]。Hong等[26]通过协同组装软模板(CTAB)和硬模板(SiO2),成功将活性Pt3Co纳米颗粒负载在传质通道周围而没有占据任何孔道内部空间,从而形成传质通道开放、活性位点大量暴露的Pt3Co/C-O(O表示开放通道)催化剂[图6(a)]。为了证明Pt3Co/C-O催化剂在孔结构设计上的优势,采用常规的后负载方法制备了一个对照样品Pt3Co/C-B(B表示堵塞的孔)催化剂。电化学测试结果表明[图6(b)],在0.9 V时,Pt3Co/C-O催化剂的质量活性为1.04 A/(mgPt),分别是商业Pt/C(0.19 A/(mgPt))和Pt3Co/C-B(0.10 A/(mgPt))的5.4和10.4倍。在H2-O2单电池中[图6(d)],Pt3Co-O催化剂能达到的峰值功率密度为1.33 W/cm2,在H2-Air单电池中可以达到0.47 W/cm2[图6(e)]。利用课题组自行设计的“拨浪鼓”电极[图6(c)]测试发现,Pt3Co/C-O催化剂在连续运行4000 s后输出相当稳定,输出电位没有明显降低,在10 mA/cm2电流密度下的电位是在15 mA/cm2时的1.55倍。而Pt3Co/C-B的输出电位在10 mA/cm2逐渐减小,表明催化剂层逐渐被水淹。通过比较10 mA/cm2时的输出电位,Pt3Co/C-O的传质效率比Pt3Co/C-B的传质效率高约29%,这归因于双孔道策略所形成的传质通道是完全开放的,从而有利于氧气和水的传输。
图6

图6 (a) Pt3Co/C-O 和Pt3Co/C-B催化剂的合成示意图; (b) ORR极化曲线以及质量活性(MA)和比活性(SA)比较;(c)催化剂以10/15 mA/cm2电流密度恒电流放电时的计时电位曲线;(d) 催化剂在H2-O2和(e)H2-Air燃料电池中的极化曲线和功率密度曲线[26]
Fig.6 (a) Schematic illustration of the preparation of Pt3Co/C-O and Pt3Co/C-B catalysts; (b) ORR polarization curves and specific activity and mass activity; (c) Chronopotentiometry curves recorded at 10/15 mA/cm2, the catalyst was loaded on the “rattle-drum” working electrode; Polarization curves and power density of H2-O2 (d) and H2-Air (e) fuel cells[26]
在上述方法的基础上,Hong等[27]通过精确调控金属离子的浓度,制备得到了高达33% (质量分数) Pt载量的PtNi合金负载空心碳球催化剂H-PtNi/C,对因催化剂层较厚而造成的传质阻力增加和MEA性能变差问题,给出了很好的解决方案[图7(b)]。其中,高载量PtNi(H-PtNi/C)和低载量PtNi(L-PtNi/C)催化剂的微观结构和合金的颗粒尺寸几乎没有差别[图7(a)]。通过在三电极测试中比较发现,低载量的PtNi合金(L-PtNi/C)的质量活性和比活性分别是高载量合金的1.6和1.2倍,说明在三电极体系中低载量合金催化剂的ORR性能较好[图7(c)]。但是将制备的材料做成膜电极之后发现,在H2-O2燃料电池中,H-PtNi/C催化剂峰值功率密度达到1.51 W/cm2,明显高于L-PtNi/C(1.25 W/cm2)和Pt/C催化剂(1.08 W/cm2),其归一化后的总Pt负载功率密度为7.55 W/(mg Pt),是L-PtNi/C催化剂获得的功率密度(6.25 W/mg Pt)的1.2倍[图7(d)],同时H-PtNi/C(4.4 A/cm2)的极限电流密度高于L-PtNi/C (3.8 A/cm2),说明适当增加催化剂中Pt的负载量会使催化剂层变薄,加快质子和电子的传输速率,缩短O2和水的扩散距离,并由此提升MEA整体性能。
图7
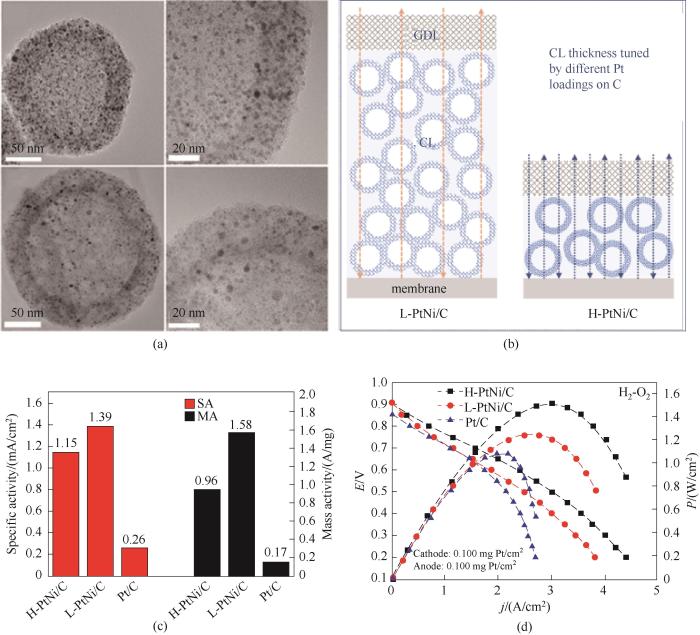
图7 (a) 高Pt (H-PtNi/C)(上图)和低Pt (L-PtNi/C)(下图)催化剂的TEM图像;(b)催化剂负载在碳(GDL为气体扩散层;CL为催化剂层) 催化剂上的膜电极(MEA)示意图;(c)催化剂在0.9 V(vs RHE)下的质量活性和比活性比较;(d)催化剂在H2-O2燃料电池中的极化曲线和功率密度曲线[27]
Fig.7 (a) TEM images of H-PtNi/C (top) and L-PtNi/C (down); (b) Schematic illustration of MEA cathodes constructed by catalysts with a high Pt loading (H-PtNi/C) and a low Pt loading (L-PtNi/C) on carbon (GDL is gas diffusion layer; CL is catalyst layer); (c) Specific activity and mass activity estimated at 0.9 V versus reversible hydrogen electrode (RHE); (d) Polarization curves and power densities of H2-O2 fuel cells[27]
2 催化剂表界面活性位构筑与介尺度结构可控制备
电化学催化是发生在催化剂表界面的电化学反应,不仅要求催化剂具有较高的导电性,还需尽可能降低催化反应的过电位,以期能最大限度降低能耗,提高反应效率。据此,如何可控构筑和优化催化剂的活性,成为电催化剂设计的关键和挑战之一[28-32]。当某催化剂引入掺杂原子、空穴以及第二相材料时,其性质从A到B的变化过程中,介域尺度内会出现区别于两者极端情况条件下的第三种主导性质——介域性质[33]。因此,关注极端情况之间的介域尺度,研究各类催化剂在结构、组分等转换过程中的介域尺度内的形成机理,以及该介域尺度结构在催化、吸附以及电荷转移过程中的非常规性质,对进一步提升催化剂的性能具有重要指导意义。
2.1 催化剂表界面活性位构筑
大量研究表明,修饰在金属氧化物上的金属纳米粒子对催化性能有显著的提高,这主要归因于独立组分之间的密切接触和电子相互作用有助于提高催化剂的本征活性,增加催化剂的利用率[34-36]。Danilovic等[37]发现用Ni(OH)2修饰金属表面,可以提高镍、银和铜催化剂在碱性溶液中的HER活性。Ni电极表面修饰了Ni(OH)2纳米团簇之后,析氢反应速率比裸露的Ni电极高4倍左右[图8(a)]。Xie等[38]报道了一种“双活性位点”MoP2/MoP纳米复合催化剂(MoP700),其中 MoP2中的Moδ+对—OH产生静电吸附,同时Pδ-对—H也产生吸附,通过双重吸附加速了水的分解,MoP相中Mo原子的充分暴露为Had的复合提供了活性位点[图8(b)]。在析氢反应中(HER),MoP700在中性溶液中表现出优异的活性和稳定性[图8(c)]。通过理论计算进一步证实了MoP700在中性pH溶液中HER活性的增强,可归因于MoP2对H2O的化学吸附和MoP对H2O/H的强结合所引起的协同作用。
图8

图8 (a) 过电位为0.3 V时的电流密度对比[37];(b)中性pH条件下,MoP700表面HER过程示意图;(c)HER极化曲线及Tafel斜率大小比较[38]
Fig.8 (a) Current densities at η=0.3 V of different metals[37]; (b) Schematic representation of the neutral pH HER on MoP700; (c) HER polarization curves and Tafel plots[38]
Jiang等[39]研究发现当TiO2负载Ru催化剂时[图9(a)],Ru-TiO2界面形成的Ru—O键时可调节Ru簇的氧化程度,使界面Ru原子的4d轨道处于半充满状态,得到的界面部分氧化Ru簇可以不吸附H2O和OH,仅选择性地促进H2的解离与氧化。当Ru团簇部分限域在TiO2晶体的晶格中时[40],金属Ru纳米颗粒和TiO2较为匹配的晶格常数使Ru 纳米颗粒稳定限域在TiO2的晶格中[图9(b)],导致金属-金属键的形成以及电子从TiO2转移到Ru金属上。电子的转移极大地改变了Ru的电子结构,并最终改变了表面物种的吸附行为,使Ru簇不仅具有超高的氢氧化(HOR)活性,还表现出了出乎意料的CO耐受性。同样地,TiO2负载的Pt基催化剂也存在类似的现象[图9(c)][41]。Pt-TiO2界面的Pt—O和Pt—Ti键可有效调控表面的抗氧化性、H的吸附和溢流,从而改变其催化活性。
图9

图9 (a)IO-Ru-TiO2 /C、Ru-TiO2 /C和Ru/C催化剂的合成示意图[39];(b)Ru@ TiO2的HAADF-STEM图像[40];(c)Pt/O–TiO2 和 Pt/Ti–TiO2上H吸附能和H,OH,H2O的吸附自由能[41]
Fig.9 (a) Schematic diagram of the synthesis of IO-Ru–TiO2/C, Ru–TiO2/C and Ru/C catalysts[39]; (b) HAADF-STEM images of Ru@ TiO2[40]; (c) H adsorption energy (ΔE*H) and free energy (ΔGads) of H, OH and H2O on Pt/O–TiO2 and Pt/Ti–TiO2[41]
在这些研究基础之上,Li等[42]再次结合理论计算指导实验证明了界面不同的化学键会产生不同性质的界面电子,其通过电荷转移改变催化剂的表界面电子结构,从而影响活性。首先利用理论计算构建了分别具有界面Ir-O,Ir-Mo及Ir-Mo/O键的负载型催化剂模型催化剂Ir/Mo-MoO2,Ir/O-MoO2和Ir/MoO2[图10(a)]。计算发现, Ir/O-MoO2催化剂中,因O更强的电负性,Ir-O界面键具有定域性,界面Ir更易接受表面的剩余电荷,使表面电荷密度降低,表面d带中心下移,从而削弱H和OH的吸附,提高了HOR本征活性。Ir/Mo-MoO2催化剂中界面Ir-Mo金属键具有更多的离域电子。界面离域电子从Mo转移给表面Ir,增加表面电荷密度,使表面d带中心上移,增强了H和OH在催化剂表面的吸附,表现出较低的HOR本征活性。而Ir/MoO2催化剂中界面Ir-Mo/O键的同时存在,促使界面产生的离域和定域电子互相抵消,导致其电子结构和物种吸附强度的不规律变化,HOR本征活性最低[图10(b)]。在理论计算指导的基础之上,在不同气氛下退火,分别制备出了表面富Mo、富O和Mo/O共存的MoO2载体,并通过液相还原将Ir负载在MoO2载体上,制备出具有不同界面化学键的Ir/Mo-MoO2,Ir/O-MoO2和Ir/MoO2催化剂[图10(c)]。电化学测试证实,催化剂在0.1 mol/L KOH溶液中的HOR活性与理论计算的HOR本征活性一致,即Ir/O-MoO2 > Ir/Mo-MoO2 > Ir/MoO2[图10(d)]。催化剂的交换电流密度分别为1.96、1.47和1.10 mA/(cm2 ECSA)。其中,Ir/O-MoO2的交换电流密度超过了之前报道的大多数Ir基催化剂。
图10

图10 Ir/Mo-MoO2,Ir/O-MoO2和Ir/MoO2在H2饱和的0.1 mol/L KOH溶液中的(a)差分电荷密度;(b) pCOHP和PDOS曲线;(c)Ir 4f XPS高分辨光谱;(d)HOR极化曲线[42]
Fig.10 (a) Charge density difference; (b) Projected crystal orbital Hamilton overlap population (pCOHP) curves and partial density of states (PDOS) of interfacial bonds;(c) Ir 4f XPS spectrum; (d) Polarization curves of investigated catalysts for HOR in H2-saturated 0.1 mol/LKOH solutions of Ir/Mo-MoO2, Ir/O-MoO2, and Ir/MoO2[42]
上述介域尺度的界面效应也存在于金属/化合物、氧化物/化合物以及负载型金属等各类含有相界面的复合催化剂中[43-47]。据此,Peng等[48]合成了高效的双功能NiO-Ni3S2/NF催化电极,通过构筑NiO与Ni3S2的相界面来同步提升催化剂的析氢与析氧活性。不同比例Ni3S2和NiO的NiO-Ni3S2/NF电极以及DFT计算进一步证实两相界面均为HER和OER(析氧反应)的最优活性位,且活性界面区含量受组分的相对含量影响,活性界面区域越大,催化性能就越高[图11(a)]。另一项工作中,他们通过在Fe3O4上进行原位硫化构建了Fe3O4/FeS2异质结构高活性单金属铁催化剂[49]。其中,通过控制硫化程度可调节界面非均质的含量[图11(b)]。高分辨率TEM图像证实纳米级的Fe3O4和FeS2晶面相互交叉键合形成了Fe3O4/FeS2异质结构,且Fe3O4/FeS2界面区域暴露在催化剂表面[图11(d)]。用X射线光电子能谱(XPS)方法研究了不同硫化程度下元素结合的演化过程。高分辨率XPS光谱显示,从Fe3O4/FeS2-1到FeS2,Fe 2p3/2峰呈先正后负的趋势,其中Fe3O4/FeS2-2.5具有最高的结合能。同样,Fe3O4/FeS2-2.5的S 2p3/2峰的结合能明显高于Fe3O4/FeS2-5和FeS2。O 1s谱图显示,Fe3O4/FeS2-2.5的Fe—O键的结合能最低。这些结果表明Fe和S的某些电荷转移到O物种上,导致了界面位置上电荷的重新分布。在碱性OER体系下,获得的Fe3O4/ FeS2-2.5催化剂具有最丰富的异质界面,表现出超低过电位和良好的耐久性[图11(c)]。密度泛函理论计算表明,界面区电荷的重新分布降低了生成含氧中间体的活化势垒,大大加速了反应动力学。
图11
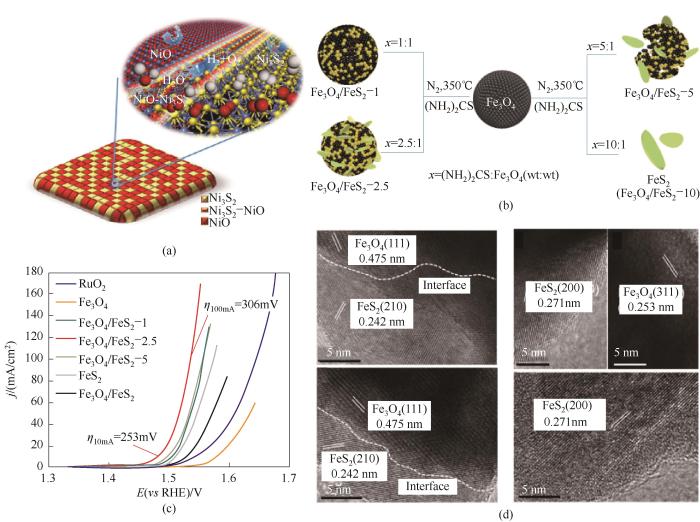
图11 (a)NiO-Ni3S2异质结及其表面催化电解水过程示意图[48];(b)Fe3O4/FeS2-x合成示意图;(c)OER极化曲线;(d)Fe3O4/FeS2-1和Fe3O4/FeS2-2.5的HRTEM图像[49]
Fig.11 (a) The schematic diagram of the monometallic NiO-Ni3S2 heteronanosheets and the water electrolysis process occurred on its surface[48]; (b) Illustration of the preparation of Fe3O4/FeS2-x samples; (c) OER polarization curves; (d) HRTEM images for the samples of Fe3O4/FeS2-1 and Fe3O4/FeS2-2.5[49]
2.2 电催化剂介尺度结构的可控制备
电化学催化反应路径通常十分复杂。研究表明,不同大小的金属物种(块状金属、纳米颗粒、单原子)在各种多相催化反应中表现出不同的催化行为(图12)。随着纳米粒子尺寸的减小,表面原子的暴露比例明显增加,同时表面原子结构、电子结构和表面缺陷也受到了调节[50-53]。然而,在纳米粒子减小的过程中,其在催化反应过程中的稳定性出现急剧下降。因此保持催化剂在纳米颗粒尺度,即处在中间区域,可以使其在具有最优活性的同时维持一定的稳定性。
图12
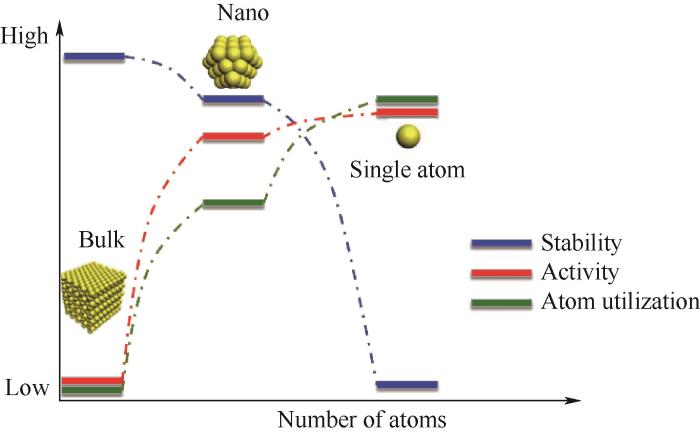
图12 电催化剂介尺度结构的可控制备
Fig.12 Controllable preparation of mesoscale structure of electrocatalyst
贵金属催化剂以其优良的活性、选择性及稳定性而倍受重视,但是贵金属的资源稀缺性决定了其价格昂贵,同时贵金属独特的物理化学性质又决定了其在多种催化反应中的不可替代性。由于催化反应总是在催化剂表面发生,暴露在表面的贵金属原子越多,贵金属利用效率就越高。因此,如何提高贵金属的原子利用效率一直是催化剂制备科学的核心问题之一。Wang等[54]发展了一种基于“空间限域”和“柯肯达尔效应”的中空Pt基催化剂通用合成方法。在H2和高温作用下,利用二氧化硅的固形和空间限域作用,控制Pt和Fe原子的迁移速度(柯肯达尔效应),使Pt原子在表面富集,Fe原子在内部富集,实现了从单组分实心Pt到中空PtFe合金的可控转化。电化学测试结果表明,在0.9 V下,其质量活性达到0.993A/(mg Pt),较商业化Pt/C催化剂提升了5.15倍。在上述方法的基础上,Zou等[55]通过精确调控吸附金属离子含量和反应温度,发现该方法不仅可以合成中空结构,还可以合成实心结构有序Pt基合金,并提出了基于化学有序能和表面偏析能主导的催化剂结构演变机理(图13)。通过热驱动的界面扩散合金化路线,允许Pt纳米粒子(NPs)在碳(Pt/C)上直接演变成无序中空PtFe合金或结构有序的实心PtFe合金纳米颗粒。并采用薄层多孔氮掺杂碳(NC)壳层原位封装PtFe合金纳米颗粒。ORR活性测试中,有序实心PtFe合金具有最佳的ORR性能,其质量活性和比活性分别为1.48 A/(mg Pt)和2.03 mA/cm2,与商用Pt/C催化剂(0.22 A/(mg Pt) 和0.31 mA/cm2)相比,提高了6.55倍和6.73倍。30000圈老化测试发现,有序PtFe合金比空心无序PtFe合金和商用Pt/C催化剂更稳定。其中,坚固的NC壳层不仅有效地防止了Pt基纳米颗粒的脱离、迁移和聚集,而且还允许电解质更平稳地进入Pt表面,从而使催化剂能够很好地保持催化活性。
图13
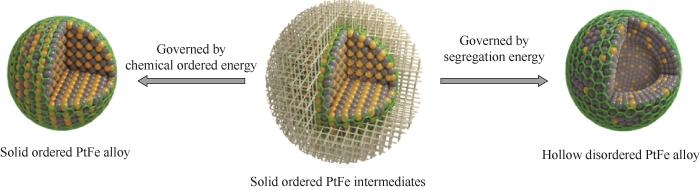
图13 基于化学有序能和表面偏析能主导的催化剂结构演变机理[55]
Fig.13 Mechanism of catalyst structure evolution based on chemical ordered energy and surface segregation energy [55]
经过科学家们十几年的努力,Pt在催化剂中的用量显著减少,然而其成本仍然是大规模商业化的主要障碍。开发基于非贵金属或无金属形式的催化剂可以克服成本问题和在成本和性能之间找到平衡的理想方法[56-58]。Hong等[59]报道了一种原位固相聚合制备单分散铁纳米颗粒嵌入氮掺杂碳气凝胶催化剂[图14(a)]。通过在水热反应中精确控制聚合度,形成含Fe2+的低聚物溶液。然后在80℃下用液氮快速固化,使低聚物能很好地保持溶液中的均匀分布。随后,在冻干条件下,低聚物在冰块的密闭空间中原位聚合成三维高交联网络。这一过程不仅有效地避免了聚合中间体的直接堆积,而且也防止了金属纳米颗粒的团聚。最终退火处理后,可使单分散的Fe/Fe3C纳米粒子嵌入氮掺杂碳气凝胶FeNC-x材料中。在Fe含量最高时,金属纳米颗粒形成了最丰富的界面区域,创造出最多的活性位点,具有最优的ORR催化活性。在碱性溶液中,半波电位高达0.919 V,0.9 V下的动力学电流密度为7.83 mA/cm2,明显优于商用Pt/C催化剂[图14(b)、(c)]。
图14

图14 (a)气凝胶结构FeNC材料制备示意图;(b)ORR极化曲线;(c)半波电位和动力学电流密度比较[59]
Fig.14 (a) Schematic illustration of the preparation of the aerogel structured FeNC materials; (b) ORR polarization curves;(c) Comparison of half wave potential and dynamic current density [59]
自1976年Jahnke等[60]通过高温热处理金属大环得到的金属氮碳催化剂(M-N/C)可以提高ORR的活性和稳定性以来,M-N/C催化剂的开发已成为非贵金属催化领域重要的研究内容之一[61-63]。He等[64]将聚吡咯(PPy)薄层包覆在铁掺杂咪唑酸沸石骨架(Fe-ZIF)上,通过界面诱导合成高活性铁氮(FeNx)富集碳纳米管(FeNx-CNTs),用于氧还原反应[图15(a)]。所构建的聚合物/MOF界面在热活化过程中抑制了Fe原子位点的团聚,从而促进了碳基体中具有FeNx配位的碳纳米管(CNTs)的形成。所制备的FeNx-CNTs催化剂在阴离子交换膜燃料电池和质子交换膜燃料电池中表现出优异的活性[图15(b)、(c)],峰值功率密度分别高达1.15和1.16 W/cm2。
图15
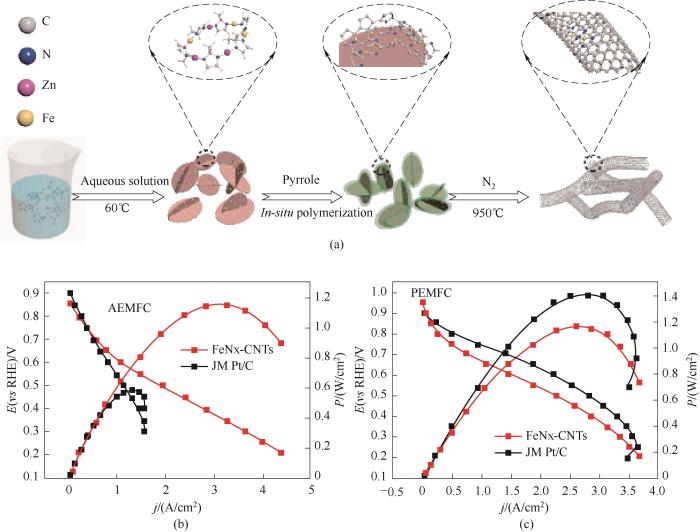
图15 (a) FeNx-CNTs催化剂的制备示意图;(b)阴离子交换膜(AEMFC)和(c)质子膜(PEMFC)H2-O2燃料电池的极化曲线和功率密度曲线[64]
Fig.15 (a) Schematic illustration of the preparation process for the FeNx-CNTs catalysts; (b),(c) Polarization curves and power densities of H2-O2 AEMFC and PEMFC [64]
3 隔膜的介尺度结构调控
碱性阴离子交换膜燃料电池(AEMFC)由于可采用成本更低的非贵金属催化剂,且氧还原动力学更快,腐蚀问题较少而受到越来越多的关注。阴离子交换膜(AEM)是AEMFC的重要组成部分,它将燃料(阳极)与氧化剂(阴极)分离,同时提供离子传输通道。在微观尺度上,隔膜距离电化学反应界面较远;而在宏观尺度上,隔膜与电极密切接触。因此隔膜的介尺度结构调控[65],对于隔膜本身和电池整体性能均有重要影响。
目前AEM主要存在两个问题:(1)OH-迁移率低,导致AEMs离子电导率也较低;(2)因为强碱性环境会加速AEM中阳离子的降解,因此AEM在高温和强碱性条件下的稳定性较差[66-69]。提高AEM离子电导率的传统方法是增加离子交换容量(IEC,即AEM中固定离子基团的浓度)。然而,高的IEC值往往伴随着膜的高维膨胀,导致AEM的力学稳定性恶化[70-72]。通过调控聚电解质的交联程度,达到平衡尺寸溶胀度和离子导电性的目的[73]。Zeng等[74]采用该方法合成了交联季铵化聚氯乙烯/交联聚乙烯醇IPN AEM,并对其结构和性能进行了系统研究。TEM图像[图16(a)]表明IPN结构中形成了高效的离子通道。测试发现,IPN AEM的OH-电导率达到141.7 mS/cm,IEC值适中,并且可以准确调控力学性能,以满足AEMFC运行的要求。使用非贵金属 (FeNx-CNTs) 作为ORR催化剂,在背压为0.2 MPa的H2-空 (无CO2) AEMFC中,峰值功率密度达到0.64 W/cm2[图16(b)]。
图16
图16 (a)低倍和高倍阴离子交换膜图像(IPN AEM);(b)碱性膜(AEMFC)H2-空燃料电池的极化曲线和功率密度曲线[74]
Fig.16 (a) Low and high magnification TEM images of IPN AEM; (b) Polarization curves and power densities of H2-air AEMFC [74]
为了进一步增加AEM在高温和强碱条件下的化学稳定性,Zeng等[75]合成了一种基于互穿聚合物网络(IPN) 结构的阴离子交换膜(IPN AEM)。首先合成了季铵化乙烯基苄基-N-甲基哌啶(QVBMP)单体,再将单体与二乙烯基苯在聚乙烯醇溶液中原位聚合,形成半IPN膜。最后,通过聚乙烯醇分子与戊二醛交联,将半IPN膜转化为全IPN膜。电镜图显示了IPN结构[图17(a)],其中一个离子导电性聚合物网络与另一个非导电性聚合物网络相互交错,形成了高效的“离子通道”。在这种结构中,IPN AEM的OH-电导率较高,IEC值适中。将IPN AEM在80℃条件下浸在6 mol/L NaOH溶液中测定膜的碱性稳定性[图17(b)],1248 h后IPN AEM仍然可以保留81%的原始电导率。在H2-O2燃料电池测试中,以非贵金属 (FeNx-CNTs) 作为ORR催化剂,背压为0.1 MPa时,峰值功率密度可以达到1.20 W/cm2。
图17

图17 (a)互穿聚合物网络阴离子交换膜(IPN AEM);(b)IPN AEM的碱性稳定性测试[75]
Fig.17 (a) Interpenetrating AEM; (b) Alkaline stability tests of IPN AEM[75]
4 结论与展望
综上所述,对于电化学催化体系中的介尺度现象和介尺度行为,本文综述了三个方面的内容:(1)多孔电极中从微孔-介孔-大孔中介尺度孔道的调控与构筑,将微孔表界面的电化学催化与大孔中物种的扩散传质有机关联起来,对提高活性位的利用率,强化传质扩散起到了重要的作用;(2)催化剂表界面晶体结构、晶面、掺杂物种以及相界面之间的相互协同,利用电荷转移、应变效应以及复合成键等方式,促使处于中间状态的介尺度区域呈现截然不同的催化性能,该介尺度特性为优化和设计催化剂提供了新的思路和方向;(3)作为电子绝缘体和离子导体的隔膜或离子交换膜,通过交联技术加速了离子扩散传输的同时,有效抑制了副反应和杂离子的扩散传输,可以进一步提高自身稳定性,协调电池两级反应。
处于介尺度下的纳米团簇、单原子等催化剂具有优异的催化活性,但其因自由能高,难以稳定存在,制备困难,成为高效宏量制备介尺度催化剂的主要挑战。对此,有三种可能的解决思路:(1)利用具有较大比表面积 (如沸石、MOF和二维材料)和丰富的锚定位点(如微孔、表面缺陷、表面官能团)的载体,增加活性位点和载体之间的作用力,有效提高纳米负载型催化剂的稳定性;(2)增加金属键之间的作用力,例如制备具有特定原子排列的有序合金,减缓金属元素,尤其是过渡金属在电催化反应中的析出,极大程度上维持催化剂本身的结构,并由此大幅提升合金纳米团簇的稳定性;(3)改变配位环境,增强金属原子与配体之间的作用力。
因此,从介尺度视角确定多孔电极、催化剂以及隔膜结构的形成机理与定向调控,为深入解释电化学催化机制以及揭示各部分之间耦合作用,量化组分、结构、尺度和反应物种间的相互影响提供了有效的手段。

- 我用了一个很复杂的图,帮你们解释下“23版最新北大核心目录有效期问题”。
- 重磅!CSSCI来源期刊(2023-2024版)最新期刊目录看点分析!全网首发!
- CSSCI官方早就公布了最新南核目录,有心的人已经拿到并且投入使用!附南核目录新增期刊!
- 北大核心期刊目录换届,我们应该熟知的10个知识点。
- 注意,最新期刊论文格式标准已发布,论文写作规则发生重大变化!文字版GB/T 7713.2—2022 学术论文编写规则
- 盘点那些评职称超管用的资源,1,3和5已经“绝种”了
- 职称话题| 为什么党校更认可省市级党报?是否有什么说据?还有哪些机构认可党报?
- 《农业经济》论文投稿解析,难度指数四颗星,附好发选题!
- 期刊知识:学位论文完成后是否可以拆分成期刊论文发表?
- 号外!出书的人注意啦:近期专著书号有空缺!!

