 0373-5939925
0373-5939925 2851259250@qq.com
2851259250@qq.com

Fe5C2-MnO x 尺度调控及催化合成气制烯烃性能研究
化工过程一般为包含多重尺度的复杂系统,大到介于生产设备和生态园区间的工厂尺度,中到介于单颗粒和反应器整体间的聚团和气泡尺度,小到介于原子、分子和整个催化剂颗粒之间的表界面和内孔尺度,这些尺度上都呈现典型的复杂性,但都可以视为基本单元相互作用形成的系统整体行为与功能[1]。在催化剂可控合成及催化反应过程中,同样涉及多个尺度,催化剂的可控制备涉及宏观合成方式与单个催化剂颗粒的孔结构、表面积及表面原子结构间的尺度调控,而催化剂构效关系研究又涉及催化剂整体催化性能与其表面无数活性位结构间的尺度调控。相对于单金属催化剂,由双金属组成的催化剂因其具有更复杂的结构特性,使其性能影响关联较大,尺度关系更为复杂[2],对其反应机制的研究涵盖了分子模拟技术、相变动力学、微观结构分析等[3-6]。
基于费托合成的合成气催化转化直接制低碳烯烃(syngas to olefins,STO)属于多相催化聚合过程,为控制目标产物选择性,第Ⅷ族金属如Fe、Co、Ru和Ni基催化剂得到广泛研究[7-11],助剂在费托合成中同样有着重要作用。Mn作为结构助剂可以促进Fe的分散,防止催化剂因积炭过多而失活[12]。MnO x 在Fe-Mn催化剂提升CO转化率的同时也降低了甲烷的选择性[13]。除此之外,MnO x 助剂能够调控产物烯烃分布,提升低碳烯烃比例[14]。Fe-Mn氧化物的制备方法对STO反应有着重要作用,可通过调变合成参数改变Fe-Mn前体化合物结构进而对其催化性能产生影响[15]。Fe-Mn氧化物在催化STO反应前需要进行还原活化,生成活性物种铁碳化合物,不同结构Fe-Mn前体对应还原活化后催化剂结构及催化性能会存在差异[16],对高催化性能Fe-Mn催化剂的可控合成可通过调变合成参数改变催化剂前体结构,进而活化生成更多铁碳化合物来实现,而该过程涵盖了从催化剂合成方式到催化剂前体结构的尺度调控及催化剂还原活化到催化剂表面活性结构的尺度调控。
铁碳化合物中Fe2C、Fe7C3和Fe5C2均对STO反应具有活性,其中以Fe5C2性能最佳,关于铁碳化合物及Fe5C2的研究包括结构分析,制备方法探究,Fe5C2对费托合成催化性能的影响及铁基纳米颗粒尺寸、分布和晶相组成与构效关系研究[17]。铁基催化剂表面铁碳化合物及活性位结构的调控需要通过宏观条件的调变来实现,包括前体合成策略的改变及还原活化过程结构演变分析等。
本文中同组成的Fe-Mn氧化物(Fe与Mn摩尔比为1∶4)分别由四种合成方法制备:低温共沉淀、沉积沉淀、前体混合煅烧及机械混合。继而通过物理吸附(BET)、X射线衍射(XRD,in situ XRD)、拉曼(Raman)、程序升温还原(TPR)等表征手段对不同合成方法得到的Fe-Mn催化剂表面性质、体相晶体结构及还原活化过程中的结构动态演变进行分析,并关联其催化STO反应性能,对Fe-Mn催化剂合成方法、表面及体相晶体结构、还原活化历程、铁碳化合物的形成及催化性能间的关系进行系统研究,为通过尺度调控得到高性能Fe-Mn催化剂提供新的策略。
1 实验部分
1.1 催化剂的制备
低温共沉淀制备方法如下:将0.04 mol硫酸亚铁和乙酸锰溶解于60 ml乙二醇中(Fe与Mn摩尔比为1∶4),恒温-10℃低温搅拌,匀速滴加浓度0.4 mol·L-1的KHCO3溶液200 ml,沉淀反应结束后,恒温-10℃持续搅拌1 h,陈化12 h,用去离子水将沉淀产物洗至中性,40℃干燥,400℃煅烧4 h,得催化剂样品,命名为GC。
沉积沉淀包括两种方式:(1)铁基沉积沉淀,先通过低温沉淀得到纯相铁沉淀物,再与乙酸锰混合至乙二醇中,后续步骤与低温沉淀法相同,所得催化剂命名为CC-Fe;(2)锰基沉积沉淀,先通过低温沉淀得到纯相锰沉淀物,再与硫酸亚铁混合至乙二醇中,后续步骤与低温共沉淀法相同,所得催化剂命名为CC-Mn。
前体混合煅烧:分别通过低温沉淀制备纯相铁及锰沉淀物,去离子水洗涤并烘干后,机械混合并煅烧,所得催化剂命名为JH-hd。
机械混合:分别通过低温沉淀制备纯相铁及锰沉淀物,去离子水洗涤、烘干并煅烧后,机械混合,所得催化剂命名为JH-dh。制备过程如图1所示。
图1

图1 4种合成方法制备过程示意图
Fig.1 Schematic diagram of four synthesis methods
1.2 催化剂的表征
催化剂的形貌采用荷兰FEI公司生产的场发射扫描电子显微镜(Quanta FEG250)采集分析(加速电压3.0 kV,工作距离5.1 nm)。样品比表面积、孔结构以及孔径由Micromeritics ASAP 2460在-196℃下测得(样品测试前在150℃下进行抽真空预处理8 h)。样品离线XRD由德国Bruker D8 advance A25型X射线仪测定,设定管电压40 kV,管电流40 mA,扫描步长0.03°,扫描范围为10°~80°。原位XRD测试流程:样品还原气氛为10%CO/Ar(体积分数),总流量50 ml·min-1,还原过程以5℃·min-1程序升温,从室温升至350℃后恒温5 h,升温过程中每50℃分析一次样品物相结构,350°C恒温时每30 min分析一次样品物相结构。样品H2-TPR采用英国HIDEN Catlab测试,分析时取100 mg样品放置于石英管中,在Ar(30 ml·min-1)气氛下,控制温度为150℃吹扫60 min。待降至室温后,将气氛换为10% H2/Ar(体积分数),在此气氛下吹扫30 min后,以10℃·min-1升温至设定温度750℃。
1.3 催化剂性能评价
采用固定床反应器对催化剂性能进行评价,测评前,将催化剂粉末压片并筛分至120~180 µm,称取0.1 g与0.1 g 120~180 µm凹凸棒粉混合装入反应管中。还原活化:反应器以10℃·min-1的升温速率从室温升至350℃,在350℃、0.5 MPa下还原5 h(体积分数为10%CO/Ar)。还原结束后,通反应气(体积分数为75%H2/CO),其中反应温度为280℃,GHSV=30000 ml·(g cat)-1·h-1,反应压力1.5 MPa。气相产物进入GC2060气相色谱仪进行在线检测。通过相关方程计算CO转化率和产物选择性等性能数据。
铁时空收率FTY(mol∙(g Fe)-1∙s-1)的计算公式为
式中,Qin、Qout为单位时间内进口、出口的总流量,ml·min-1;νCO为进口气的CO浓度;XCO为CO的转化率;vi 表示出口气体中各组分的体积分数;Ci 表示出口气体中各组分的碳数;
2 结果与分析
2.1 催化剂物相结构表征
为考察不同合成方法Fe-Mn催化剂前体晶相组成,对纯Fe、纯Mn、低温共沉淀、沉积沉淀、前体机械混合的样本进行XRD分析,结果如图2所示。纯Fe沉淀物相结构主要为Fe2O3,纯Mn沉淀物相主要为MnCO3。低温共沉淀得到的Fe-Mn催化剂前体在24.2°、31.4°、37.5°、41.4°、45.2°和51.6°处出现了MnCO3(JCPDS No.44-1472)的衍射峰,没有新的特征峰与Fe物种有关联,原因应是铁物种高度分散于锰晶格中或者Fe沉淀晶体尺寸太小(小于5 nm)未被检测出[18]。对比MnCO3衍射峰位置发现,共沉淀晶体衍射峰位置细微向右偏移,由于Mn离子半径比Fe大,Fe掺入Mn晶格导致晶格收缩,发生晶格畸变,形成FeMn固溶体结构。而沉积沉淀法制得的样品衍射峰整体向左偏移,根据布拉格衍射定律nλ=2dsinθ,θ变小,d值变大代表了晶格膨胀,说明掺入了比主体原子半径大的杂原子,归因于二次沉淀中的晶粒无法以细小离子形式渗入已形成沉淀的大晶体晶格中,CC-Fe样品因Fe已沉淀无法以单一离子形式进入锰晶格中或者以团聚的大颗粒状态与Mn掺杂,导致衍射峰整体向左偏移,CC-Mn样品因Mn已沉淀完全Fe离子已经无法均匀掺杂于Mn晶格中,大多只能以沉淀形式沉积于表面。
图2
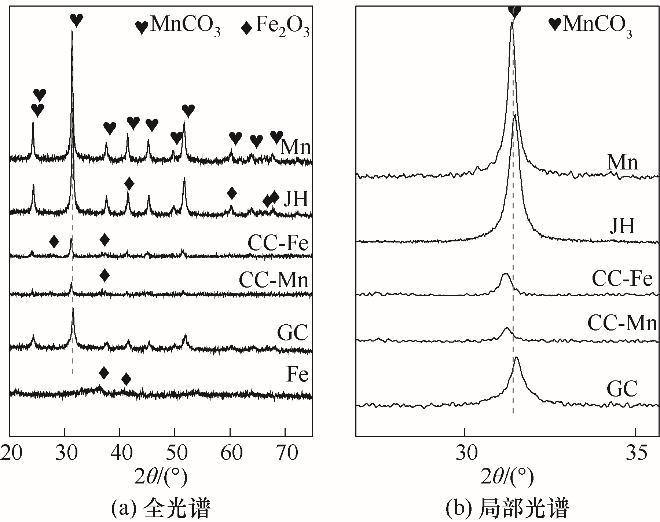
图2 不同合成方法Fe-Mn催化剂前体XRD谱图
Fig.2 XRD patterns of Fe-Mn precursors prepared by different methods
前体经400℃煅烧之后,物相发生变化。如图3所示,共沉淀法制得的催化剂主要以锰基氧化物MnO2、Mn2O3为主,其中37°、42°、55°、67°分别对应MnO2的(101)、(111)、(211)和(310)晶面(PDF#24—0735),该方法中未观察到Fe物种的衍射峰,与前体物相表现一致,推测是添加的铁含量过低或Mn的加入改善了α-Fe2O3的分散性[19]。对于沉积沉淀以及机械混合法制得的催化剂因其无法在合成状态下形成均匀的固溶体结构,明显观察到有关α-Fe2O3的衍射峰。
图3

图3 不同合成方法Fe-Mn催化剂XRD谱图
Fig.3 XRD patterns of Fe-Mn catalysts prepared by different methods
从不同合成方法制备的Fe-Mn催化剂XRD表征中看出,催化剂前体以及煅烧后,共沉淀法制得的催化剂主要是与锰相关的衍射峰,而其他方法制得的催化剂铁、锰衍射峰共存。显然,共沉淀法制备的Fe-Mn催化剂,Fe物种在催化剂中分散度最好。
不同合成方法制备的Fe-Mn系列的催化剂织构性质列于表1中,低温共沉淀法制备得到的催化剂有着较高的比表面积(158.61 m2∙g-1)以及较小的粒径尺寸(37.83 nm),结合Fe-Mn催化剂的XRD分析结果,低温共沉淀得到的Fe-Mn催化剂结构更为疏松。为了进一步研究孔结构,对催化剂进行了吸脱附曲线分析,如图4所示,不同合成方法得到的Fe-Mn催化剂在较高的相对压力(P/P0 = 0.5~0.9)下均表现出明显的滞后环,在较低的P/P0下吸附非常低,说明样品具有介孔性[20],通过孔径分布也可得到,不同合成方法催化剂孔径分布在6~10 nm之间。
表1 Fe-Mn催化剂的织构性质
Table 1
| 催化剂 | 比表面积/(m2∙g-1) | 孔容/ (m3∙g-1) | 孔径/nm | 平均尺寸/nm |
|---|---|---|---|---|
| Fe(纯铁) | 91.04 | 0.29 | 8.69 | 65.91 |
| GC | 158.61 | 0.33 | 6.03 | 37.83 |
| CC-Fe | 120.29 | 0.27 | 6.62 | 49.88 |
| CC-Mn | 147.98 | 0.36 | 7.19 | 40.55 |
| JH-hd | 124.47 | 0.27 | 6.45 | 48.21 |
| JH-dh | 98.76 | 0.30 | 9.19 | 60.76 |
| Mn(纯锰) | 102.31 | 0.27 | 7.61 | 58.64 |
新窗口打开| 下载CSV
图4

图4 不同合成方法Fe-Mn催化剂吸脱附曲线及孔径分布
Fig.4 Adsorption and desorption curves and pore size distribution of Fe-Mn catalysts prepared by different methods
为了观察不同合成方法Fe-Mn微球催化剂的结构和形貌,对其进行了SEM表征,如图5所示。不同合成方法对催化剂的形貌、织构、粒径和团聚程度有显著影响。由共沉淀法制备的催化剂是由大量细小均匀、结构松散的颗粒组成。两种沉积沉淀法表现出不同的结构特点:CC-Fe优先对Fe2+进行沉淀形成晶体,Mn2+沉淀与Fe沉淀晶粒复合形成絮状大颗粒形式,而CC-Mn优先对Mn2+进行沉淀形成球状颗粒,继而Fe2+沉淀煅烧形成棒状氧化物附着在球状锰沉淀晶体表面。而前体混合后煅烧及Fe、Mn两样品煅烧后单纯机械混合均表现为大颗粒团聚。基于图6中各样品粒径分布结果,低温共沉淀Fe-Mn催化剂宏观球体粒径集中分布在400 nm左右,基于沉积沉淀法制备的CC-Fe及CC-Mn宏观粒径较小(100 nm),前体混合煅烧样品宏观粒径分布在200 nm左右,而煅烧后简单机械混合的样品粒径分布在100~500 nm之间。结合铁锰共沉淀的XRD、BET及SEM分析结果,当Fe2+与Mn2+共沉淀时,Fe2+沉淀形成的粒径极小的颗粒进入Mn2+沉淀形成的MnCO3晶体内部通过物理作用力引发MnCO3晶体发生分裂生长,并使其最终形成了新颖的束状形貌的沉淀物,使其保留MnCO3晶体结构的同时获得了较高的比表面积[21]。
图5

图5 不同合成方法Fe-Mn催化剂SEM图
Fig.5 SEM images of Fe-Mn catalysts prepared by different methods
图6

图6 粒径分布
Fig.6 Particle size distribution
Fe-Mn催化剂的分子振动信息通过Raman光谱进行测试,该测试采用532 nm激光光源对不同合成方法制备得到的催化剂进行离线分析,如图7所示,这些样品均在639 cm-1处出现非常明显的峰,属于MnO2中Mn—O的拉伸振动模式[22]。共沉淀法制备得到的Fe-Mn催化剂出现三个主要振动吸收峰,在305 cm-1、678 cm-1均归属为Mn2O3的振动吸收峰[23],189 cm-1归属为MnO2的振动吸收峰[24];沉积沉淀和机械混合法制得的催化剂则在639 cm-1出现了MnO2的振动吸收峰,314 cm-1、383 cm-1对应于Mn2O3振动吸收峰,并且CC-Mn以及机械混合两种催化剂在210~220 cm-1,270~280 cm-1之间出现了α-Fe2O3的主要振动吸收峰,这与XRD表征结果一致。从不同合成方法制备的Fe-Mn催化剂Raman光谱图中明显可观察到:共沉淀法得到的Fe-Mn催化剂谱峰宽化(639~678 cm-1),这是由于该方法下的催化剂粒径变小[25],Fe掺杂使晶格发生畸变所致,且未观察到有关Fe的振动吸收峰,表明Fe离子很好进入Mn晶格中,这很好地对应了XRD、SEM表征结果;CC-Fe制备的催化剂有着较高的Mn—O键的振动吸收峰,而Fe—O键的振动吸收峰相对较低,表明二次沉淀Mn后,有较多的Mn晶粒沉积在Fe的表面,因Fe含量较少,所以有较多的Mn的振动吸收峰被发现;CC-Mn制备的催化剂有着较高的Fe—O键的振动吸收峰,而Mn—O键振动吸收峰较低,这是由于先沉淀Mn后沉淀Fe晶粒,导致Mn晶粒先沉淀,而Fe离子已无法进入锰晶格中,以Fe沉淀的形式沉积于表层,所以有较高强度的Fe相关的振动吸收峰被发现;机械混合法制备得到的Fe-Mn催化剂则同时有着Fe和Mn相关的振动吸收峰。
图7
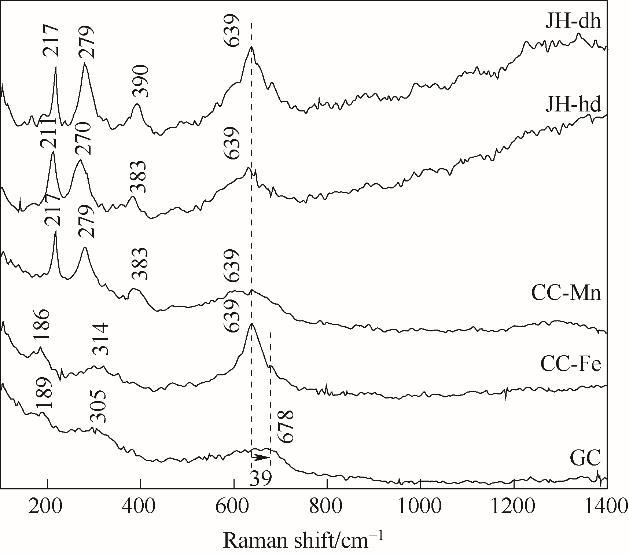
图7 不同合成方法Fe-Mn催化剂的拉曼光谱图
Fig.7 Raman spectra of Fe-Mn catalysts prepared by different methods
2.2 催化剂活化过程研究
不同合成方法制备的Fe-Mn催化剂H2-TPR结果如图8所示,共沉淀法制备得到的Fe-Mn催化剂211~251℃的还原峰可归属于MnO2和Mn2O3还原为Mn3O4,其氧化锰还原峰较小是由于催化剂合成过程中,Fe物种进入Mn沉淀晶体晶格内部,相互影响,使得有较少的氧化锰被还原;与之类似的是CC-Mn催化剂的氧化锰还原峰,表明Fe2+与Mn2+共沉淀或者Mn2+预先沉淀,Fe物种都能进入锰氧化物沉淀晶体晶格内部,与Mn物种形成稳定的晶体结构;而对于CC-Fe催化剂,锰氧化物还原峰较强,峰型与机械混合Fe-Mn催化剂的锰氧化物还原峰类似,是由于在合成过程中Fe2+先进行了沉淀,形成粒径较大的颗粒,在后续Mn2+沉淀过程中,无法进入Mn沉淀晶体晶格内部,故该还原峰体现的是Mn2+沉淀形成的锰氧化物的本征还原峰,该结果与不同合成方法得到的Fe-Mn催化剂的Raman分析结果相一致;不同合成方法Fe-Mn催化剂第二个还原峰360℃左右处可归属为Fe2O3和Mn2O3的混合氧化物还原成(Fe1-yMn y )3O4物相,而(Fe1-yMn y )3O4物相继续还原成(Fe1-x Mn x )O物相的过程,由于Fe-Mn的相互作用,改善了Fe3+结合到MnO晶格中并稳定了方铁矿相[18],也难以进一步还原。从谱图中可看出,沉积沉淀以及机械混合法更容易还原为方铁矿相,生成了较多合金相。图中机械混合法在高温500℃左右出现微弱耗氢峰,代表Fe3O4还原为FeO,说明未掺杂到Mn晶格中Fe的还原行为,与Raman谱图相佐证。
图8
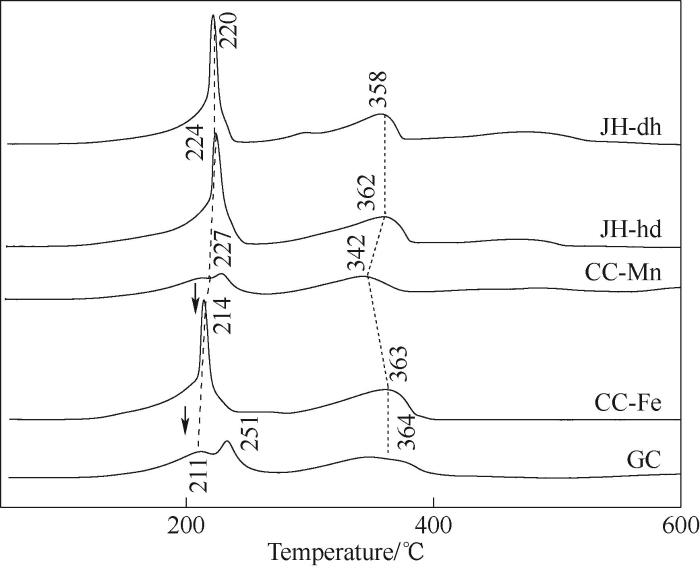
图8 不同合成方法Fe-Mn催化剂的H2-TPR谱图
Fig.8 H2-TPR spectra of Fe-Mn catalysts prepared by different methods
为研究Fe-Mn催化剂还原活化过程中的结构演变,对Fe-Mn催化剂的还原活化过程进行了原位XRD分析,活化气氛为10% CO/Ar(体积分数),活化首先在室温以5 ℃·min-1的升温速率将催化剂升温至350℃,继而恒定350℃活化5 h,其物相结构演变如图9、图10所示。共沉淀制备得到的催化剂在室温下有着明显的锰氧化物结构α-Mn2O3,随还原温度升高至200℃时,Fe3O4相开始出现,Fe-Mn催化剂中Fe物种逐渐被还原为Fe3O4;温度到达350℃时,Fe5C2特征峰开始出现,同时出现了少部分的铁锰氧化物的衍射峰以及Fe3C物种的衍射峰。沉积沉淀法及机械混合法制备的Fe-Mn催化剂在室温下均为α-Fe2O3和α-Mn2O3物相,当温度升至300℃时,物相变为Fe-Mn复合氧化物,且有较少的积炭峰出现,从350℃开始,Mn3O4和Fe3O4逐渐转变为铁锰复合氧化物,同时出现了少量的铁碳化合物的特征峰,由于复合氧化物有着较高的峰强,削弱了原有晶格中Fe—O的部分振动形式[26],结合H2-TPR分析结果,沉积沉淀以及机械混合法得到的Fe-Mn催化剂更易还原成(Fe1-x Mn x )O复合相,进一步阻止还原过程。
图9
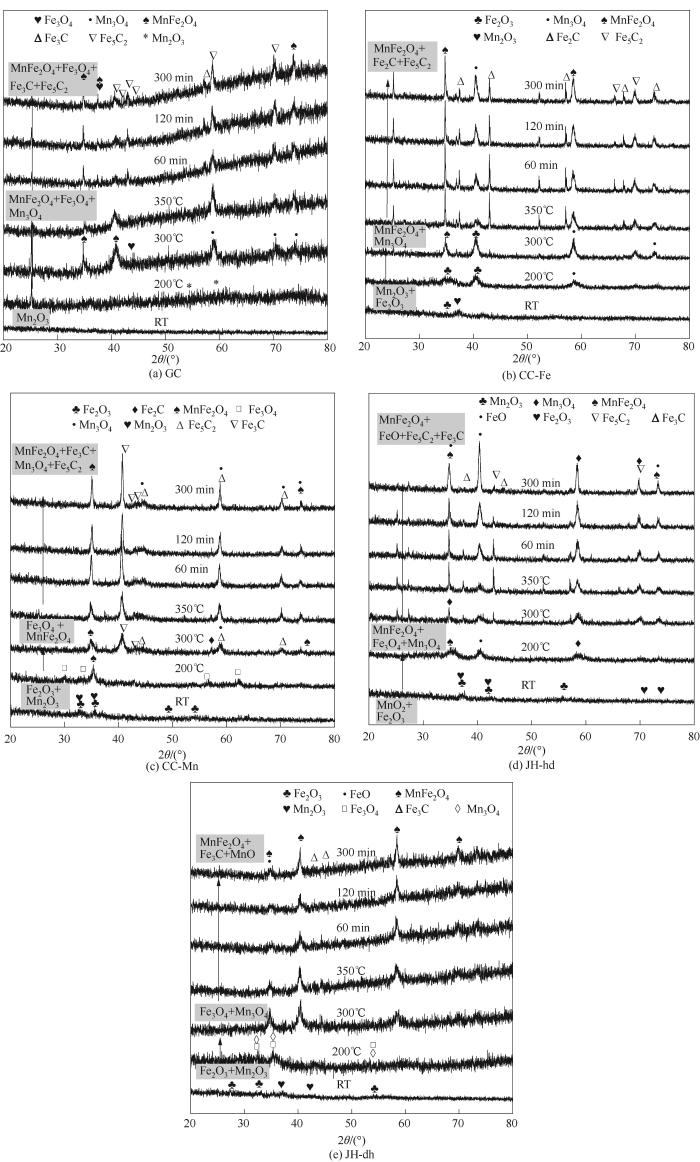
图9 10% CO/Ar还原过程中Fe-Mn催化剂的原位XRD分析
Fig.9 In situ XRD patterns of Fe-Mn catalyst during reduction at 10% CO/Ar
图10
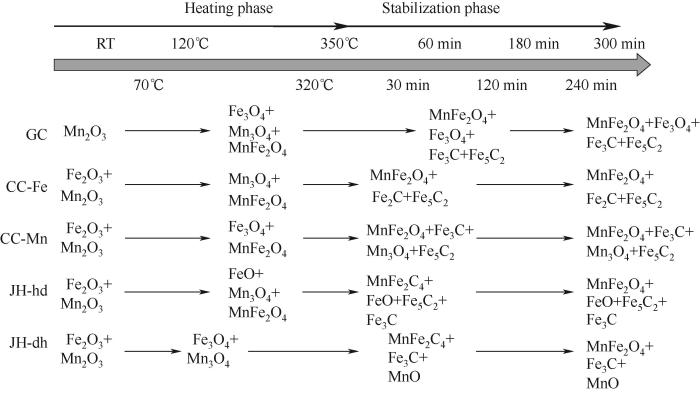
图10 不同合成方法Fe-Mn催化剂结构演变过程
Fig.10 Structural evolution of Fe-Mn catalysts prepared by different methods
根据Fe-Mn催化剂原位XRD谱图,对Fe-Mn催化剂还原活化过程中各物相含量变化及铁碳化合物晶粒尺寸变化进行分析,结果如表2、表3所示(MnFe2O4、Fe5C2、Fe3C、Fe2C和MnO相对晶相含量为100%)。Fe-Mn催化剂催化活性主要取决于催化剂粒径尺寸和孔隙结构主导的比表面积,以及催化剂表面铁碳化合物(如α-Fe5C2, γ-Fe3C)的比例[27]。表2表明不同合成方法对应的铁碳化合物含量有所不同,晶粒尺寸也有较大差别。GC催化剂表现出较高的Fe5C2含量(55.2%)以及较小的晶粒尺寸(19.69 nm),有利于催化剂提高催化活性及多产低碳烯烃;CC-Fe催化剂表现出高的Fe3C含量(62.0%)以及较小的晶粒尺寸(16.82 nm),高的Fe3C含量可以促使催化剂多产长链烯烃,同时JH-hd催化剂也表现出高的Fe3C含量(69.8%),但晶粒粒径相对CC-Fe的较大(21.68 nm)。
表2 不同合成方法Fe-Mn催化剂的晶相含量
Table 2
| 催化剂 | 晶相含量①/% | ||||
|---|---|---|---|---|---|
| MnFe2O4 | Fe5C2 | Fe3C | Fe2C | MnO | |
| GC | 17.9 | 55.2 | 26.9 | — | — |
| CC-Fe | 17.1 | 15.9 | 62.0 | 5.0 | — |
| CC-Mn | 31.7 | 20.3 | 31.7 | — | 16.3 |
| JH-hd | 20.9 | 9.3 | 69.8 | — | — |
| JH-dh | 37.9 | — | 38.6 | — | 23.5 |
① 根据各物相在XRD所占主要峰面积进行Jade估算。
新窗口打开| 下载CSV
表3 不同合成方法Fe-Mn催化剂的晶粒尺寸
Table 3
| 催化剂 | 晶粒尺寸①/nm | |
|---|---|---|
| Fe5C2 | Fe3C | |
| GC | 19.69 | 28.44 |
| CC-Fe | 20.80 | 16.82 |
| CC-Mn | 26.68 | 27.12 |
| JH-hd | 27.14 | 21.68 |
| JH-dh | — | 26.79 |
① 根据Scherrer公式D=Kλ/(βcosθ) 估算其晶粒尺寸。K为常数;λ为X射线波长;β为衍射峰半高宽;θ为衍射角。
新窗口打开| 下载CSV
综合不同合成方法制备得到的Fe-Mn氧化物的结构分析及还原活化过程的结构演变分析,可以通过对合成方法的调变来实现对催化剂前体结构的尺度调控,且由于不同催化剂前体在活化演变历程中生成的活性相、活性相尺寸都不同,进而通过还原活化也实现了对催化剂表面活性物种结构的尺度调控。这一结果也证实了催化体系间各尺度间的关联,催化剂宏观合成条件实时影响催化剂的前体结构,而催化剂前体结构也实时影响还原活化后的催化剂表面活性物种结构。
2.3 催化剂性能测试
不同合成方法制备的Fe-Mn催化剂STO反应性能数据如表4所示。由表可知,催化活性与其制备方法密切相关。共沉淀法制备得到的Fe-Mn催化剂催化性能较高(20.07%的CO转化率),沉积沉淀法制备的催化剂催化性能居中(14%~15%的CO转化率),而机械混合制备得到的催化剂则表现出较低的催化活性(4%~8%的CO转化率)。
表4 不同合成方法Fe-Mn催化剂的STO性能数据汇总
Table 4
| 催化剂 | CO conversion/% | FTY① | CO2 selectivity/% | Hydrocarbon distribution②/% | α | O/P③ | |||
|---|---|---|---|---|---|---|---|---|---|
| CH4 | C5+ | ||||||||
| GC | 20.07 | 4.37 | 6.37 | 13.28 | 17.45 | 38.60 | 24.29 | 0.54 | 2.32 |
| CC-Fe | 15.49 | 2.77 | 22.97 | 17.55 | 14.48 | 20.11 | 19.71 | 0.71 | 1.47 |
| CC-Mn | 14.36 | 2.23 | 33.08 | 9.49 | 14.19 | 27.95 | 15.28 | 0.53 | 2.25 |
| JH-hd | 8.41 | 1.55 | 20.84 | 26.13 | 13.44 | 23.62 | 15.97 | 0.58 | 1.99 |
| JH-dh | 4.63 | 0.89 | 17.23 | 21.56 | 14.14 | 26.08 | 20.98 | 0.55 | 2.04 |
①铁时空收率(FTY)为单位时间单位质量Fe将CO转化为烃类产物的能力(10-5 mol CO∙(g Fe)-1∙s-1)。②碳氢化合物的分布以碳为基础进行归一化。③C2~7的烯烃与烷烃比值。
新窗口打开| 下载CSV
对于共沉淀法制备的催化剂,其
图11

图11 Fe-Mn催化剂催化STO反应性能分析
Fig.11 Catalytic performance of the Fe-Mn catalysts for STO reaction
由于催化剂的催化性能取决于其还原活化后的表面微观结构,GC催化剂具有较高的催化活性和烯烃选择性,可以归结为以下两个方面:(1)粒径小且均匀的铁碳化物;(2)碳层的限制效应。碳层可以防止催化剂的烧结,因此粒径较小的碳化物表现出较高的催化活性和稳定性,虽然碳层不可避免地会覆盖一些活性位点,导致初始阶段的活性相对较低,但也会促进铁碳化物的形成[33]。如图11所示,随着反应时间的延长,GC催化剂表面生成大量碳化物,使其低碳烯烃选择性逐渐提高。
3 结 论
本文研究了不同合成方法对Fe-Mn催化剂活化过程结构演变及催化STO反应性能的影响,结论如下。
(1)对不同合成方法得到的Fe-Mn催化剂前体物相结构进行了XRD、BET、SEM及Raman表征,结果表明,共沉淀法制备的Fe-Mn催化剂由于在合成过程中,Fe2+可以进入Mn2+沉淀形成的晶体内部,造成晶格畸变,使最终合成得到的Fe-Mn催化剂比表面积高,晶粒尺寸小。
(2)对Fe-Mn催化剂进行H2-TPR及活化过程的原位XRD分析,结果表明沉积沉淀和机械混合法制备的Fe-Mn催化剂因还原成较多的(Fe1-x Mn x )O物相,导致较低的还原渗碳程度,而共沉淀法制备的Fe-Mn催化剂还原后生成最多量的Fe5C2,且对应晶粒尺寸也最小,使其催化STO反应表现出最高的CO转化率(20.07%)和低碳烯烃选择性(38.60%)。
上述研究结果为指导合成高催化性能Fe基催化剂提供了新的策略,即通过调控合成方法来实现高比表面积、高分散度Fe基催化剂的尺度调控,进而通过还原活化以实现对高Fe5C2含量及低晶粒尺寸的Fe基催化剂的尺度调控,最终使其在催化STO反应中表现出高的CO转化率及低碳烯烃选择性。

- 我用了一个很复杂的图,帮你们解释下“23版最新北大核心目录有效期问题”。
- 重磅!CSSCI来源期刊(2023-2024版)最新期刊目录看点分析!全网首发!
- CSSCI官方早就公布了最新南核目录,有心的人已经拿到并且投入使用!附南核目录新增期刊!
- 北大核心期刊目录换届,我们应该熟知的10个知识点。
- 注意,最新期刊论文格式标准已发布,论文写作规则发生重大变化!文字版GB/T 7713.2—2022 学术论文编写规则
- 盘点那些评职称超管用的资源,1,3和5已经“绝种”了
- 职称话题| 为什么党校更认可省市级党报?是否有什么说据?还有哪些机构认可党报?
- 《农业经济》论文投稿解析,难度指数四颗星,附好发选题!
- 期刊知识:学位论文完成后是否可以拆分成期刊论文发表?
- 号外!出书的人注意啦:近期专著书号有空缺!!

