 0373-5939925
0373-5939925 2851259250@qq.com
2851259250@qq.com

羟基磷灰石-氧化石墨烯复合微球的制备及性能
羟基磷灰石(hydroxyapatite,HA)又称碱式磷酸钙,化学式为Ca(5 PO4)(3 OH),是牙齿和骨骼的主要无机组成成分,因HA具有良好的生物相容性、生物活性和生物降解性,在医疗材料领域应用广泛[1-2].另外,由于HA具有离子交换特性,在废水处理方面也有广泛的应用[3-4].HA可降解吸收,对人体基本不存在毒副作用,因此可作为药物载体材料使用[5].但是HA作为生物材料单独使用时脆性较大,且韧性较低[6],为HA提高的力学性能,目前主要是将HA与其他高分子材料进行复合[7-8].
氧化石墨烯(graphene oxide,GO)是石墨烯的衍生物,其单层碳原子构成的二维空间的基面连接有大量亲水性官能团,可以与其他的官能团进行酰化和酯化等共价反应[9],具有超强吸附能力,对亲/疏水药物可以高负载.同时,在生物环境中,GO具有良好的相容性、分散性、较高的载药量和良好的缓释特性等特点,能有效解决药物易团聚和药效短等问题[10].
姜黄素(curcumin,Cur)是植物界很稀少的具有二酮的色素,其形态为橙黄色结晶粉末,味稍苦,不溶于水,可溶于乙醇和冰醋酸,常作为食用色素使用.有研究证明,姜黄素具有降血脂、抗肿瘤、抗炎、利胆和抗氧化等作用[11].姜黄素的分子结构中有两个活性酚结构,因此具有一定的抗氧化能力.作为一种天然酚性物质,Cur还含有抗肿瘤和消炎物质,且有抗氧化特性,能诱导癌细胞凋亡[12].此外,Cur通过抑制核因子-kappa B(nuclear factor-kappa B,NF-κB)通路激活[13]和降低组织内活性氧水平等方式可延缓椎间盘退变进程[14-15].但Cur在水溶液中的溶解度不高,稳定性差,而且在人体肠道内代谢快,生物利用率低,不能很好地发挥Cur的作用[16].因此,为提高Cur的稳定性与利用效率,将Cur作为目标药物模型进行载药复合微球的测试,为Cur治疗食管癌、膝关节炎、阿尔茨海默病和糖尿病等提供参考.
模板法先通过较为简单方便的途径合成模板,再通过物理或化学方法将目标产物合成到模板上,去除模板后便可得到具有模板形貌与尺寸的目标产物,分为硬模板法和软模板法[17].本研究以合成的球状碳酸钙作为硬模板,通过水热法以离子交换方式成功制备了球状中空的HA-GO复合微球,并通过X射线粉末衍射(X-ray powder diffraction,XRD)、傅里叶变换红外光谱(Fourier transform infrared spec⁃troscopy,FTIR)、拉曼红外光谱、场发射扫描电子显微镜(field emission scanning electron microscope, FESEM)和紫外可见光近红外分光光度计(ultravio⁃let visible near-infrared spectrophotometers, UV-Vis-NIR)等测试手段对合成的复合微球进行表征分析,同时对其载药和释药等性能进行研究.
1 实 验
试剂:磷酸氢二钠((NH3)2HPO4)和二水氯化钙(CaCl2·2H2O)购于上海阿拉丁试剂有限公司;碳酸钠(Na2CO3)、柠 檬 酸(C6H8O7)和 无 水 乙 醇(CH3CH2OH)购自广州市东红化工厂;姜黄素(C21H20O6,药用级别,98%)购自上海源叶生物科技有限公司;氧化石墨烯购自湖南丰化材料发展有限公司;中国仓鼠卵巢细胞株购自江苏凯基生物技术股份有限公司.
仪器:主要有日本岛津傅里叶变换红外光谱仪、德国Bruker的(D8-ADVANCE)X射线粉末衍射仪、日本电子株式会社公司的JSM-7800&TEAM型场发射扫描电子显微镜、英国雷尼绍公共有限公司的InVia Reflex型激光显微共聚焦拉曼红外光谱仪以及Micromeritics公司的全自动比表面积与孔隙度分析仪.
制备不同反应初始浓度的CaCl2溶液与Na2CO3溶液,向CaCl2溶液中加入0. 5 mol/L的柠檬酸控制剂搅拌溶解,加入质量浓度为0. 1 mg/mL的GO超声分散30 min,用1. 0 mol/L NaOH调节pH=5. 8.将Na2CO3溶液倒入CaCl2的混合溶液中,同时迅速搅拌,调节pH≈11,搅拌1 h,静置沉淀22 h.用去离子水和乙醇洗涤多次,于烘箱60~70℃干燥,制得CaCO3-GO模板.根据CaCl2溶液与Na2CO3溶液的反应初始浓度共设4组实验,每组实验中c(CaCl2)=c(Na2CO3),依次为0. 1、0. 3、0. 5和1. 0 mol/L,获得的CaCO3-GO模板对应记作0. 1CaCO3-GO、0. 3CaCO3-GO、0. 5CaCO3-GO和1. 0CaCO3-GO.
分别称取4种不同初始浓度下制备的CaCO3-GO样品各1. 0 g,各自加入到50 mL去离子水中,再加入0. 06 mol/L(NH4)2 HPO4搅拌溶解,用1. 0 mol/L NaOH调节溶液pH≈11.于180℃下将混合溶液在反应釜中反应,控制水热反应时间.待反应结束后,水洗3~5次,干燥后收集.实验对水热反应时间(分别为3、6和12 h)和不同初始浓度制备的CaCO3-GO两个因素通过正交实验进行研究分析. 4种CaCO3-GO模板生成的HA-GO产物对应标记为0. 1HA-GO、0. 3HA-GO、0. 5HA-GO和1. 0HA-GO.
依据ISO 10993. 1-2018医疗器械的生物学评估标准对载体材料进行体外细胞毒性测试,采用CCK-8法检测细胞毒性[18].称取0. 9 g样品浸泡在无水乙醇24 h,过滤干燥后紫外消毒灭菌.再将样品浸泡在20 mL的完全培养基中,37℃、CO2恒温培养浸提24 h,采用微孔滤膜过滤,收回浸提液.并以此浸提液浓度为基准划分体积分数梯度,依次为 1. 0%、 5. 0%、 10. 0%、 20. 0%、 50. 0%、70. 0%、80. 0%和100. 0%的浸提液和完全培养基9个梯度,以完全培养基作为对照组,其余作为实验组.将中国仓鼠卵巢(Chinese hamster ovary,CHO)细胞接种于96孔板,每组10孔.各组分别加入200 μL对照培养液或不同浓度的浸提液.培养72 h后,在每孔加入CCK-8试剂,轻微震荡后继续放入恒温细胞培养箱中培养,2~3 h后用实时酶联免疫检测仪测量各孔在450 nm波长处的光密度值D(4 5 0),实验重复测5次,取平均值,计算得出细胞相对增殖率.
配制一系列姜黄素无水乙醇溶液测定吸光度,计算回归方程,建立标准曲线.
分别称取4种CaCO3-GO模型生成的HA-GO样品各50 mg,将它们置于姜黄素的乙醇溶液中匀速混合24 h.在8 000 r/min下离心15 min,分离并收集上清液,重复加入无水乙醇离心,使收集到的上清液总量为20 mL.测量上清混合液的光密度,通过与之前姜黄素乙醇溶液的标准曲线对比,计算微球的载药量和包封率,并设置3个平行实验以减少误差.同时取空白样,置于无水乙醇中同样操作,排除空白微球的浸出物对测试过程的影响.包封率和载药量的计算公式为
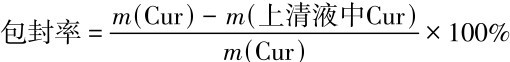
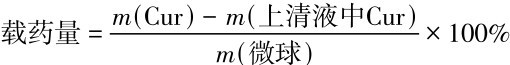
按2015版中国药典的规定对载药微球进行体外释放试验[19],释放溶液使用体积分数为20%的乙醇稀盐酸溶液来模拟人工胃液(pH=1. 5). 称取50 mg载药样品,放入透析袋中,然后放入释放溶液中,在37℃水浴条件下搅拌,进行药物缓释实验.分别于0. 5、1、2、4、8、12、24、48、72和96 h时取出4 mL释放溶液,同时补充4 mL模拟的人工胃液,测量各个时间点释放溶液的在波长为(427 ± 1)nm处吸光度,同样进行3个平行样测试,减小误差.缓冲液中药物的累积释放率E为其中, V0为缓冲溶液的总体积(单位: mL); V为每次取出缓冲溶液的体积(单位: m L ); ρn为第n次吸取缓冲溶液时测定的药物质量浓度(单位:mg/mL);ρi为第i次置换取样时测定的药物溶液质量浓度(单位:mg/mL);m (药物)为载药微球中包裹的药物质量(单位:mg) .
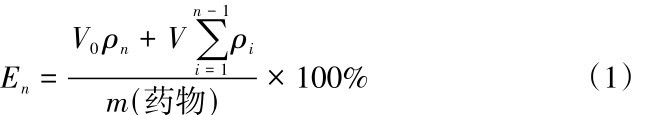
2 结果与讨论
图1为不同反应浓度条件下制备的CaCO3-GO的XRD图谱.碳酸钙主要有文石、方解石和球霰石3种无水结晶相.方解石是热力学最稳定,能量最低的晶型,而球霰石是热力学最不稳定.能量最高,文石介于两者之间[20].低反应浓度(c(CaCl2)=c(Na2CO3)=0. 1 mol/L)与高反应浓度(c(CaCl2)=c(Na2CO3)=1. 0 mol/L)得到的XRD图谱显示均为方解石晶型,根据比对方解石的标准卡片(PDF#05-0586),图谱的强衍射峰基本与方解石的(104)、(018)和(116)等晶面符合,进而可以判断合成的为方解石的碳酸钙.对比球霰石的标准卡片(PDF#33-0268),在0. 3 mol/L反应条件下,出现球霰石的衍射峰,其(112)和(114)晶面衍射峰强度很高,不过仍然显示存在方解石的衍射峰.在0. 5 mol/L时,也可以明显看出两种晶型的衍射峰同时存在,但方解石的衍射峰强度强一些.说明在低反应浓度时生成的主要是热力学较稳定的方解石晶型的CaCO3,在增大反应浓度后,部分方解石晶型向球霰石晶型转化,但浓度增大到一定程度时,存在形式又转化为稳定的方解石.说明反应浓度确实影响碳酸钙的晶型变化,在适当的反应浓度下可以生成球霰石晶型.GO的特征衍射峰在2θ=11. 7°处,可能由于本实验中强度太弱,没有检测到.
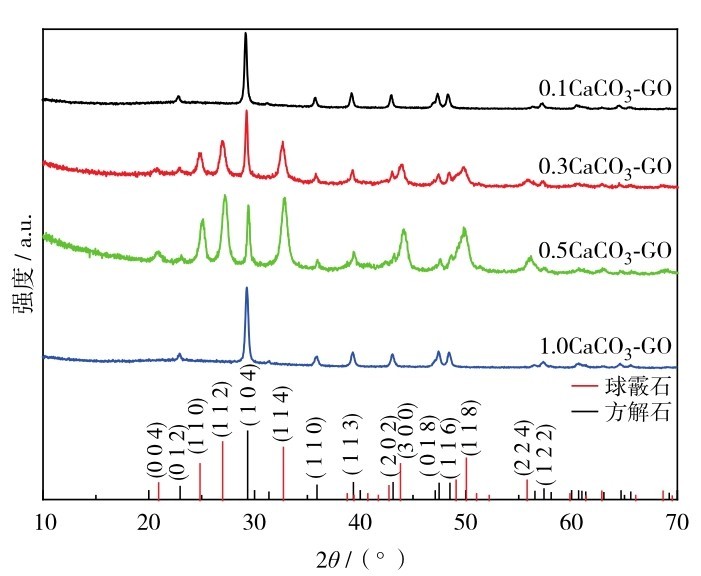
图1 不同反应浓度制备的CaCO3-GO的XRD图谱Fig. 1 The XRD patterns of CaCO3-GO prepared with different reaction concentrations.
图2为不同反应浓度度条件下制备的CaCO3-GO的FTIR图谱.由图2可见,c(CaCl2)=c(Na2CO3)分别为0. 1 mol/L和1. 0 mol/L制备的样品中在712 cm-1处有明显吸收峰,属于方解石的CO32-基团的O—C—O面内变形振动峰.而0. 5 mol/L样品在745 cm-1处出现吸收峰,这属于球霰石的CO32-基团的O—C—O面内变形振动峰,同时也在712 cm-1有明显吸收峰,说明方解石与球霰石2种晶型共存.当反应浓度为0. 3 mol/L时,712 cm-1处的吸收峰较弱,不太明显;745 cm-1处的球霰石吸收峰比较明显而且清晰,说明此样品主要是球霰石型的CaCO3,这也与上述XRD分析结果一致.
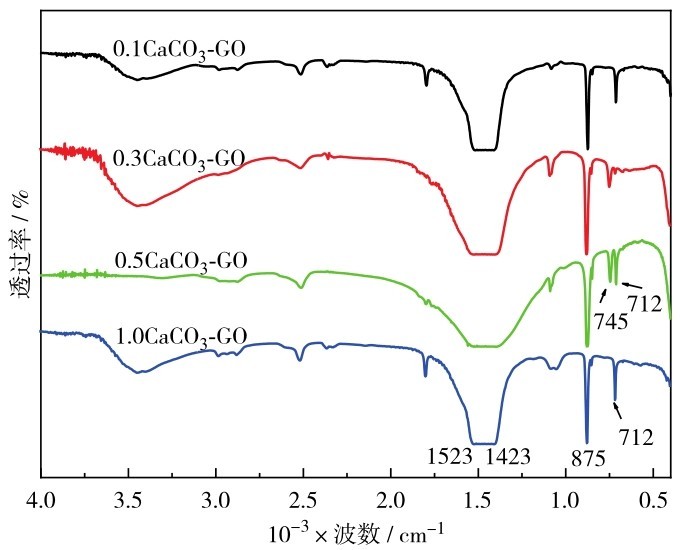
图2 不同反应浓度制备的CaCO3-GO的FTIR图谱Fig. 2 FTIR images of CaCO3-GO prepared with different reaction concentrations.
图3为不同反应浓度制备的CaCO3-GO的FESEM图像.从图3可明显看出,低反应浓度时合成的是棒状颗粒,长度为1. 0~2. 5 μm,直径在100~200 nm,呈束状聚集(如0. 1 mol/L时);当反应浓度增大到0. 3 mol/L时,出现球状颗粒,且表面光滑,球状比较完整,部分呈椭球状,夹杂少许棒状颗粒,球形的粒径为0. 9~2. 0 μm,粒径分布比较均匀;当浓度增至0. 5 mol/L时,球状减少,多呈圆饼状且大小不一,同时还有不太规则的棒状存在;当高反应浓度1. 0 mol/L时,基本上没有微米级的可辨识形貌,只有纳米颗粒呈团聚分布.从图3可明显看出,反应浓度对碳酸钙是否能呈现微球状的形貌有重要的影响.

图3 (a)0. 1CaCO3-GO、(b)0. 3CaCO3-GO、(c)0. 5CaCO3-GO和(d)1. 0CaCO3-GO的FESEM图像Fig. 3 FESEM images of (a) 0. 1CaCO3-GO, (b) 0. 3CaCO3-GO, (c) 0. 5CaCO3-GO, and (d) 1. 0CaCO3-GO.
图4为4种不同初始反应浓度制备的CaCO3-GO水热后生成的HA-GO的XRD图谱.总体来看,碳酸钙的衍射峰基本消失.与HA的标准卡片(PDF# 09-0432)对比可以看出,不同反应浓度下生成的HA-GO复合物均出现HA的特征衍射峰[21-22].说明CaCO3转化成了HA,且纯度也比较高.同时,在2θ≈11°时有很微弱的峰,此处为HA与GO重叠的衍射峰.
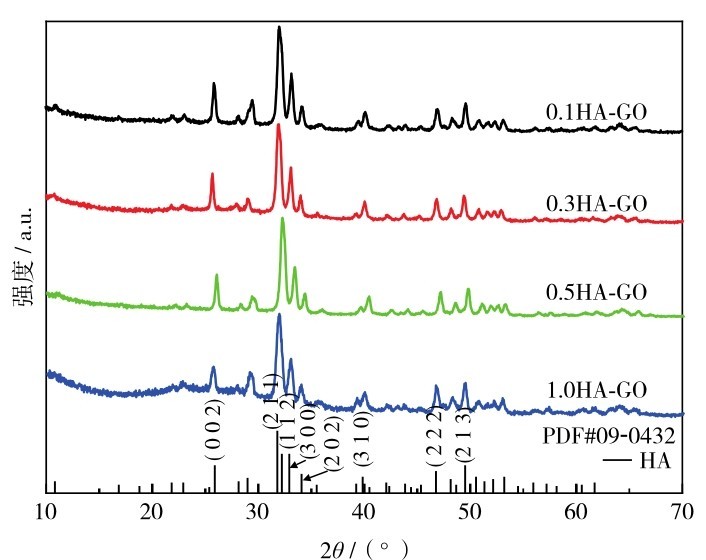
图4 不同初始反应浓度制备的CaCO3-GO水热后生成的HA-GO的XRD图谱Fig. 4 XRD patterns of HA-GO generated from CaCO3-GO prepared by different initial reaction concentrations after hydrothermal treatment.
图5为不同初始反应浓度制备的CaCO3-GO水热后生成的HA-GO的傅里叶红外图谱.由图5可见,在569和605 cm-1处为PO43-基团的弯曲振动峰, 1 050 cm-1处的吸收峰为PO43-基团的伸缩振动吸收峰[23].同时还发现,712 cm-1处的球霰石的CO32-基团振动峰已消失,745 cm-1处方解石的特征吸收峰也不再出现.1 462 cm-1和415 cm-1处为GO的C—O振动峰,也为CO32-基团的C—O振动峰,可能是由于水热反应后仍有部分CO32-存在于HA中.3 500 cm-1处的吸收峰应该是GO的—OH峰.结合上述XRD分析,碳酸钙已基本转化为HA,说明该水热反应条件下阴离子交换比较彻底.
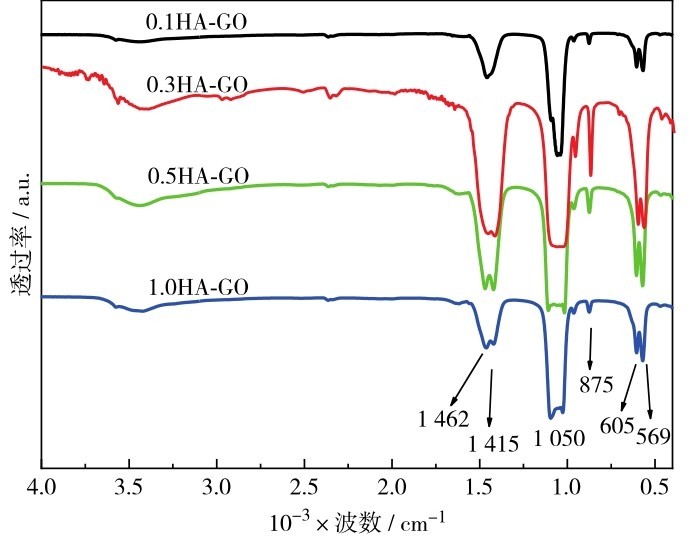
图5 不同初始反应浓度制备的CaCO3-GO水热后生成的HA-GO的傅里叶红外图谱Fig. 5 FTIR patterns of HA-GO generated from CaCO3-GO prepared by different initial reaction concentrations after hydrothermal treatment.
图6为不同反应浓度下制备的HA-GO的SEM图.在图6(a)中,0. 1CaCO3-GO经过水热反应后得到0. 1HA-GO粉末,虽然其基本形态还呈棒状,不过已转化为HA颗粒,呈散乱分布;图6(b)为0. 3HA-GO,从形貌上看基本成球状,从部分破碎的微球可以发现内部呈中空结构,微球表面十分粗糙,大小比较均一;图6(c)为0. 5HA-GO,可以看到部分微球缀连在GO的边缘;从图6(d)的高倍电镜下可以看到,微球表面有明显的颗粒存在,这是转化后的纳米HA生成在表面;而图6(e)中的1. 0HA-GO已经团聚成块状了.
图7是在180℃进行CaCO3-GO水热反应,反应时间分别为3、6和12 h生成的HA-GO的XRD图谱.由图7可见,当反应时间为3h时,对比标准卡片已经出现HA的晶相,而球霰石的特征衍射峰仍然存在,说明两者共同存在,此时CaCO3已经开始转化为HA.经过高温高压,PO43-开始跟CaCO3中的CO32-进行交换[24],但仍有CaCO3的衍射峰,说明部分CaCO3没有转化成功.而随着时间的延长,交换越来越彻底,在反应6 h和12 h时,已经没有CaCO3的衍射峰,说明CaCO3已完全转化为HA.

图6 (a)0. 1HA-GO、(b)0. 3HA-GO、(c)0. 5HA-GO、(d)16 000倍镜下的0. 3HA-GO和(e)22 500倍镜下的1. 0 HA-GO的SEM图Fig. 6 SEM images of (a) 0. 1HA-GO, (b) 0. 3HA-GO, (c) 0. 5HA-GO, (d) high magnification of 0. 3HA-GO, and (e) 1. 0 HA-GO.
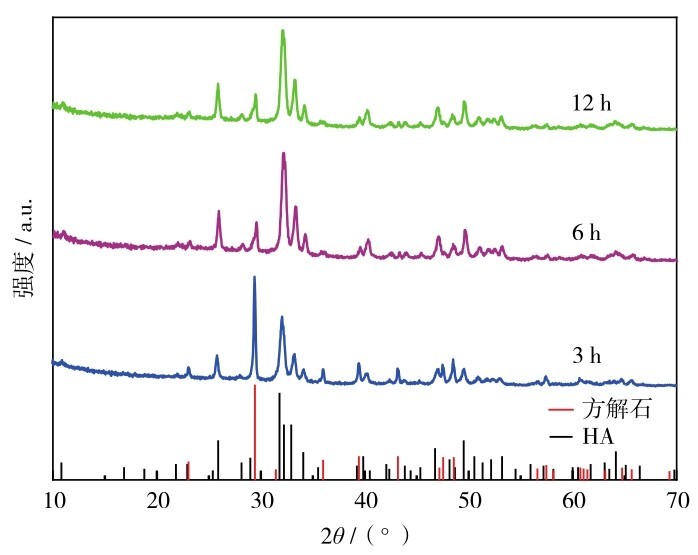
图7 不同水热时间制备的HA-GO的XRD图谱Fig. 7 XRD patterns of HA-GO prepared at different hydrothermal times.
图8为不同水热反应时间(3、6和12 h)条件下制备的HA-GO的SEM图像.由图8可以看出,样品基本都呈球状,并保持模板CaCO3的形态.水热反应时间3h的样品,微球表面粗糙,且有微小的颗粒存在.反应时间为6h时,微球更加粗糙,表面颗粒变大.增加水热时间至12 h,有的微球破碎,表面粗糙,内部呈现中空结构,有网络状微结构存在.结合XRD分析,可以看出水热反应时间为6 h时能获得形貌较好、转化彻底的HA-GO微球.

图8 水热时间分别为(a)3 h、(b)6 h和(c)12 h制备的HA-GO的SEM图Fig. 8 SEM images of HA-GO prepared with hydrothermal time of (a) 3 h, (b) 6 h, and (c) 12 h.
因此,以下实验将使用0. 3HA-GO且水热反应时间为6 h、形貌较好的复合微球进行相关测试分析.
图9为HA-GO复合微球的拉曼光谱.从图9可见,在1 350 cm-1处,GO和0. 3HA-GO都有1个明显的振动峰,称为D带,这是石墨烯的无序振动峰.位于1 600 cm-1的峰属于石墨烯的本征拉曼模式,称为G带.589和960 cm-1分别对应于v3 (PO43-)反对称变角振动峰及v1(PO43-)对称伸缩振动峰,是HA的特征峰.用D带与G带的强度比(ID/ IG)表征石墨烯材料的无序度.GO的ID/ IG=0. 85,HA-GO的ID/IG=0. 94,复合样品拥有较高的ID/IG,表明复合样品中GO的缺陷密度增加.
图 10是0. 3HA-GO微球的Barrett-Joyner-Halenda (BJH)孔径分布图以及吸脱附等温曲线.根据吸脱附等温曲线可知,微球的吸脱附等温曲线符合典型的Ⅲ型等温线,其滞后环为H3型[25],说明微球的组成微粒之间存在许多狭缝型的孔隙,观察相应SEM图也可相互印证这点.测得比表面积为22. 71 m2/g.根据孔径大小分类:孔径≤2 nm为微孔;孔径介于2~50 nm的为介孔;孔径≥50 nm为大孔.通过BJH法分析微球孔径可知,微球的平均孔径为30~50 nm,属于介孔材料.结合以上分析可知,制备的HA-GO复合微球是内部中空结构的介孔材料.
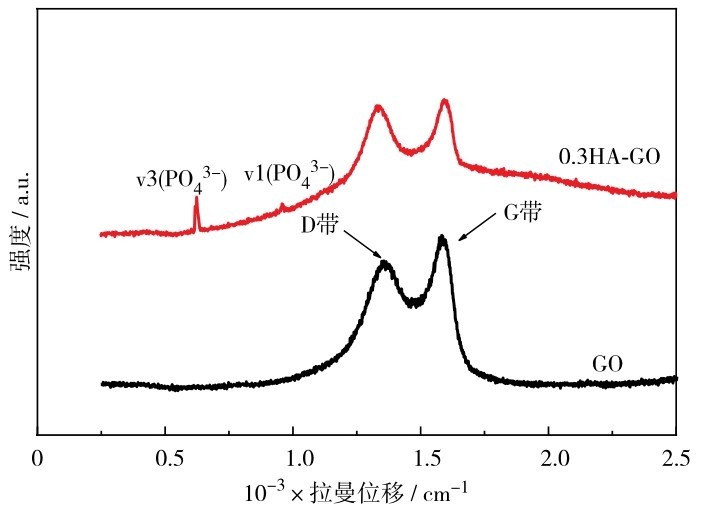
图9 GO和0.3HA-GO复合微球的拉曼光谱Fig. 9 Raman spectra of GO and 0. 3HA-GO composite microspheres.
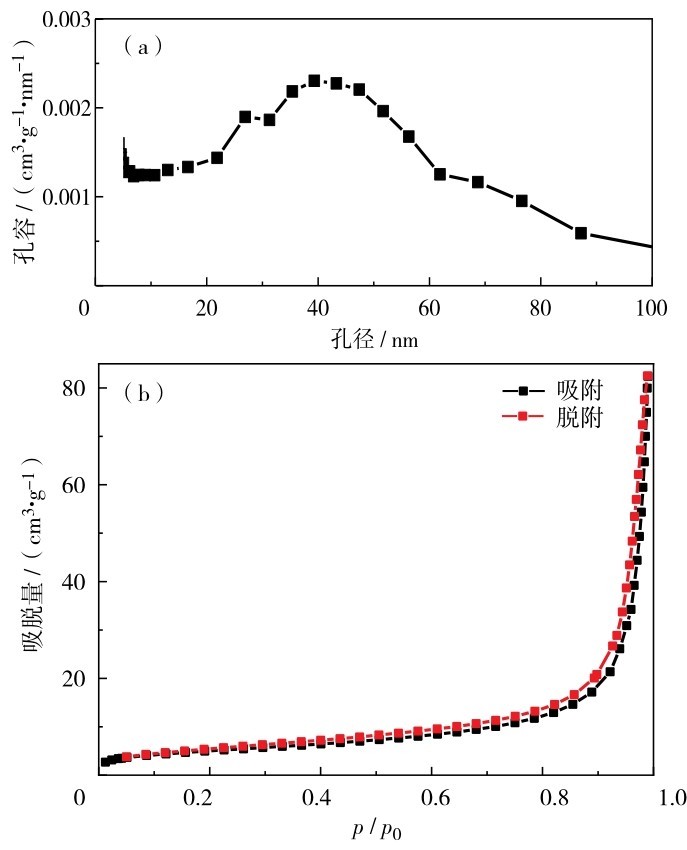
图 10 HA-GO微球的BJH孔径分布图及吸脱附等温曲线(a)BJH孔径分布;(b)吸脱附等温曲线Fig.10 Adsorption-desorption isotherm curve of HA-GO microspheres. (a) BJH pore size distribution map, (b) adsorption and desorption isotherms.
通过体外培养CHO细胞评估HA-GO复合微球对细胞增殖的影响,结果如图 11.CCK-8的结果表明,以对照组统计学数量为参考标准,不同浓度的浸提液对细胞增殖基本没有负面影响,说明含复合微球物质的培养液没有毒性,因而复合微球对细胞增殖基本上没有毒性.
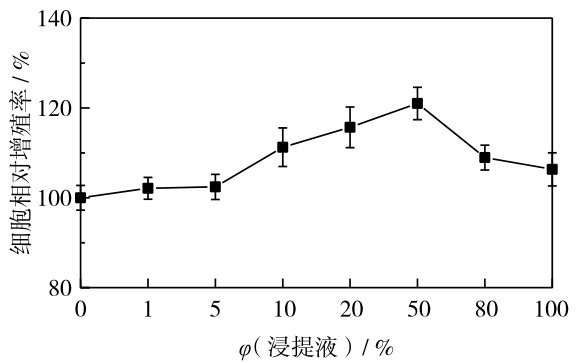
图 11 不同体积分数的微球浸提液培养下的细胞相对增殖率Fig. 11 Relative proliferation rates in culture extracts of different amounts of the microspheres.
通过紫外分光光度计测定得到姜黄素在无水乙醇中的最佳吸收波长为(427 ± 1)nm.同时,在(427 ± 1)nm波长处测定配置的一系列姜黄素乙醇溶液的吸光度,绘制标准曲线如图 12.通过线性拟合,发现在姜黄素乙醇溶液质量浓度为0. 05~2. 0 μg/mL内呈良好线性关系.
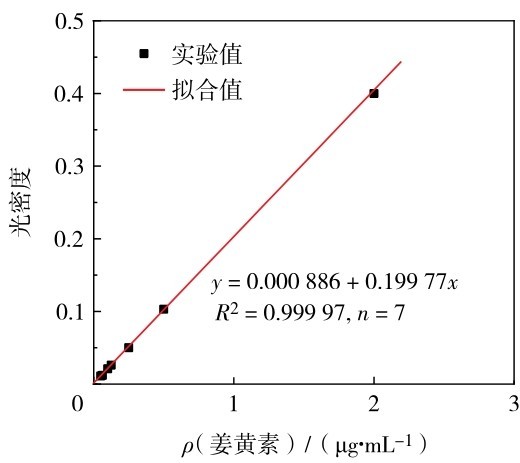
图 12 姜黄素在无水乙醇中的标准曲线Fig. 12 Standard curve of curcumin in absolute ethanol. Black square is the experimental data,and red line is the fitting curve.
包封率和载药量是衡量载药微球载药性能的重要指标,载药量和包封率高可使微球材料得到充分利用,从而减少原材料的浪费和给药次数,减轻患者频繁给药的痛苦,临床应用价值更高.图 13为复合微球的包封率及载药量,由图 13可见, 0. 3HA-GO的样品拥有较好的负载能力,其载药量达到(2. 95 ± 0. 19)% ,包封率达到(20. 90 ± 0. 31)%.这主要是因为0. 3HA-GO形成的是表面粗糙内部中空的微球,给药物提供了更多的着位点,利于药物吸附.
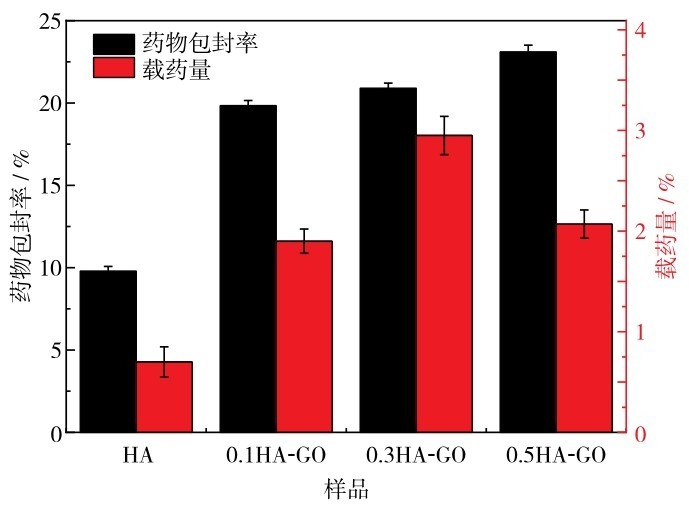
图 13 HA-GO的包封率与载药量的柱状图Fig. 13 Histograms of drug encapsulation efficiency (black) and drug loading (red) of HA-GO.
由于姜黄素属于醇溶性物质,因此使用体积分数为20%的乙醇稀盐酸溶液(pH=1. 5)作为模拟人工胃液进行体外释药测试(图 14).由图 14可以看出,载药复合微球初释放时呈平稳释放,没有明显的突释现象,在给药后120 h时的累计释放率达到72%.由曲线的走势来看,释放率还在缓慢抬升,说明药物后期还会释放,最终药物累积释放率将高于72%,说明HA-GO微球既能起到缓释,又能基本将药物完全释放.
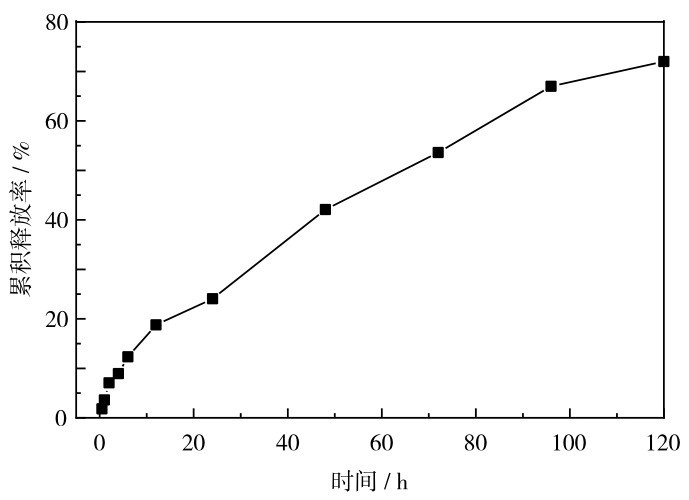
图 14 载药微球在体积分数为20%乙醇的模拟人工胃液(pH=1. 4)中的累积释放率随时间变化Fig. 14 Cumulative release rate of drug-loaded microspheres in artificial gastric juice (pH=1. 4) supplemented with volume fraction of 20% ethanol changes with time.
结 语
综上研究可知,初始底物的反应浓度很大程度上影响CaCO3-GO复合粉末的微观形貌,是CaCO3能否形成球状结构的重要影响因素.而在转化过程中,水热时间是转化进程是否完成的重要因素, CaCO3-GO复合粉末的基本微观形貌决定了转化后的HA-GO复合粉末的形貌.研究结果表明,球状碳酸钙作为硬模板,通过水热法以离子交换方式能成功制备球状中空的HA-GO复合微球.球形CaCO3-GO的最优反应条件为0. 3 mol/L的反应浓度.制备球形HA-GO的最优合成条件为:使用0. 3 mol/L的CaCl2溶液和0. 3 mol/L的Na2CO3溶液制备的CaCO3-GO复合产物,在180℃的高压反应釜中水热反应6 h.制备得到的CaCO3-GO复合微球的粒径为5. 1~7. 7 μm,孔径为30~50 nm.制备的HA-GO复合微球,以姜黄素作为药物模型进行载药性能及释放性能的测试.结果发现,球状结构的微球的确能提高药物的负载量,复合微球的包封率能达到(20. 90 ± 0. 31)% 、载药量达到(2. 95 ± 0. 19)%,对姜黄素起到了缓释作用,并且释放较为彻底.研究结果表明,HA-GO载药复合微球展现出良好的医用前景.

- 我用了一个很复杂的图,帮你们解释下“23版最新北大核心目录有效期问题”。
- 重磅!CSSCI来源期刊(2023-2024版)最新期刊目录看点分析!全网首发!
- CSSCI官方早就公布了最新南核目录,有心的人已经拿到并且投入使用!附南核目录新增期刊!
- 北大核心期刊目录换届,我们应该熟知的10个知识点。
- 注意,最新期刊论文格式标准已发布,论文写作规则发生重大变化!文字版GB/T 7713.2—2022 学术论文编写规则
- 盘点那些评职称超管用的资源,1,3和5已经“绝种”了
- 职称话题| 为什么党校更认可省市级党报?是否有什么说据?还有哪些机构认可党报?
- 《农业经济》论文投稿解析,难度指数四颗星,附好发选题!
- 期刊知识:学位论文完成后是否可以拆分成期刊论文发表?
- 号外!出书的人注意啦:近期专著书号有空缺!!

