 0373-5939925
0373-5939925 2851259250@qq.com
2851259250@qq.com

β-淀粉样蛋白的聚集及其调控
阿尔茨海默病(Alzheimer’s disease, AD)是最常见的神经退行性疾病之一,严重威胁着人类的健康,给社会和家庭带来了沉重的经济负担。据统计,世界范围内约有5000万人受AD的影响,我国约有600万的AD患者,预计到2050年全世界患病人数将达1.52亿[1]。因此亟需开发能够治愈AD的药物或手段。临床上,AD患者表现为认知能力的下降、记忆力的衰退、情绪和性格的改变、语言功能障碍等。AD的致病原因较为复杂,各种因素均能诱发AD,宏观上有年龄、基因、病毒感染、糖尿病、脑部创伤等因素;微观层面如Tau蛋白过磷酸化、淀粉样蛋白(amyloid β-protein, Aβ)的沉积、金属离子稳态失衡等[2]。其中,Aβ级联假说受到了广泛的研究和认可[3-5]。Aβ级联假说认为,细胞外Aβ病理性的沉积是引发神经损伤的关键因素。Aβ单体能够自发聚集形成纤维状的聚集体,并在其纤维化的过程中产生大量异质的寡聚体。Aβ寡聚体和纤维对神经细胞产生严重的毒性,导致突触功能受损,从而引发功能性障碍。因此,调控Aβ的聚集已成为治愈AD的关键。
近年来,在Aβ聚集及其调控方面已进行了广泛的研究[6],但由于Aβ聚集过程呈现复杂的多尺度性,目前对寡聚体结构的认识尚不充分,因此制约着Aβ聚集抑制剂的开发。为此本文综述了以Aβ聚集及其调控为核心的相关介尺度过程。首先,简要介绍Aβ的产生和聚集过程以及与介尺度科学的关系。随后,按照不同的分类标准系统总结Aβ聚集过程中产生的各种介尺度寡聚体,以及Aβ寡聚体的细胞损伤机制。然后,对目前广泛研究的Aβ抑制剂,如小分子类、多肽类、蛋白类、纳米类抑制剂进行归纳和阐述。最后,讨论AD治疗以及Aβ抑制剂目前存在的挑战和需要重点研究的方向。本综述将不仅增强对介尺度结构的共性认识,更为设计有效的Aβ聚集抑制剂乃至治愈AD提供参考。
1 Aβ的产生和聚集过程
Aβ多肽由淀粉样前体蛋白(Aβ precursor protein, APP)水解切割得到。APP存在于谷氨酸能神经元表面,含695~770个氨基酸残基,在维持突触可塑性、神经元正常的生理功能方面具有重要作用。在淀粉样生成途径中,APP经β-分泌酶和γ-分泌酶的水解,产生39~43个氨基酸残基的Aβ片段;而在非淀粉样生成途径中,由于APP会经α-分泌酶的水解,因此不会产生Aβ片段(图1)[7]。在大脑内,Aβ40占80%~90%,而Aβ42占5%~10%。由于Aβ42比Aβ40多含有2个疏水性的氨基酸,因此,Aβ42具有更高的聚集倾向。此外由Aβ42聚集所产生寡聚体的细胞毒性也比Aβ40大。在正常生理条件下,Aβ能被胰岛素降解酶和脑啡肽酶降解;未被降解的Aβ会穿越血脑屏障(blood-brain barrier, BBB)进入循环系统[8]。Aβ单体具有一定的生理活性,能保护成熟的神经元免受兴奋性毒性死亡,还能够调节电压门控钾通道。但是Aβ一旦发生聚集,会产生较大的神经毒性,诱导神经元死亡。
图1
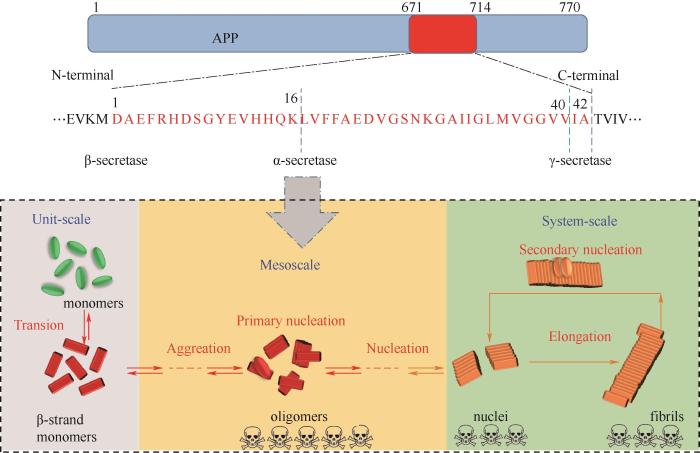
图1 Aβ产生和聚集过程中的介尺度科学问题
Fig.1 Schematic illustration of mesoscience in the production and aggregation of Aβ
研究表明,Aβ的聚集过程符合“核依赖聚集动力学”,根据表征聚集体中β-sheet含量的硫黄素T (thioflavin T, ThT)荧光实验可将聚集过程分为成核期、生长期和稳定期三个阶段[9]。聚集始发于Aβ单体的构象转变。首先是Aβ单体暴露出疏水性的基团,两个单体之间通过自身识别位点经二聚化作用结合到一起,形成反平行β-sheet结构的二聚体(dimer)[10]。二聚体不断发生构象的转变,和其他单体或者二聚体通过疏水和氢键作用组装形成亚稳态的寡聚体,并形成“聚集核”,此过程被称为初级成核(primary nucleation)过程。随后,以上述形成的“聚集核”作为单体或寡聚体结合的模板,聚集形成大量的原纤维,从而进入聚集加速的生长期。在生长阶段,聚集体的β-sheet结构快速增加。最后,进入稳定期,形成大量成熟的Aβ纤维[11]。此外,形成的原纤维或者纤维聚集体会断裂形成新的碎片,这些碎片化的纤维能作为新的“聚集核”,促进Aβ寡聚体的富集并加速Aβ的聚集,此过程被称为二次成核(secondary nucleation)过程(图1)。虽然Aβ的纤维化大致符合上述聚集路径,但是具体聚集的分子机理不仅与Aβ亚型相关,还与初始结构、溶解度等多种因素有关[12]。
上述Aβ聚集反应是包含多种结构的复杂性生物反应过程,属于介尺度科学的研究范畴。如图1所示,Aβ的聚集经历了单体的构象转换,多种不同分子量、形态结构寡聚体的形成,以及最终的成核并生长为纤维的过程。其中单体为单元尺度,核和纤维为系统尺度,而不同的Aβ中间体则是介于单元与系统尺度之间的介尺度[13]。Aβ聚集过程是多种介尺度结构竞争与协调共同作用的结果,且Aβ中间体形态的多样性、结构的多尺度性以及反应的多变性导致Aβ聚集研究的复杂性[14]。为此,亟需解析蛋白质聚集过程中耦合连接的关键介尺度结构、阐明Aβ介尺度结构的变化规律、建立Aβ聚集的介尺度动态模型,以针对性地调控关键介尺度结构的形成,从而实现Aβ聚集的调控以及AD治疗的目标。
2 介尺度Aβ寡聚体的分类及其性质
与其他复杂性系统类似,尽管Aβ聚集过程呈现出不同的层次,但每个层次的边界尺度上系统的行为相对简单,易于表征和分析[13]。如聚集的初始条件为在体内累积大量的Aβ单体,聚集的最终状态是形成富含β-sheet结构的纤维状Aβ聚集体。而在它们之间的介尺度层次上,系统性行为非常复杂,缺乏成熟的理论描述。因此对于介尺度Aβ寡聚体结构与行为的定量描述与调控是介尺度科学研究的核心问题,也是耦合Aβ聚集过程多尺度问题的关键之处。但目前的实验手段很难对Aβ聚集过程中介尺度寡聚体结构进行精确的描述,因此难以阐明介尺度结构动态形成的机理。现阶段对介尺度Aβ寡聚体的研究呈现出显著的跨学科、跨领域特征,目前主要通过生物物理学技术、成像手段、计算机模拟等方法进行模糊的描述,且结果具有很大的局限性。其中常见的分析方法有通过分子量大小进行区分的SDS-PAGE (SDS-polyacrylamide gel electrophoresis);根据Aβ寡聚体半径大小区分的尺寸排阻色谱(size-exclusion chromatography, SEC);成像技术包括电镜(electron microscopy, EM)、原子力显微镜(atomic force microscopy, AFM)成像;结构解析手段包括X射线衍射(X-ray diffraction)、固态核磁共振技术(solid-state nuclear magnetic resonance techniques, ssNMR);特异性抗体对寡聚体特定构象的识别等[7]。以下通过分子量、形貌、结构、构象等原则对介尺度Aβ寡聚体进行分类和性质总结。
2.1 按分子量分类
按分子量的不同,Aβ寡聚体可分为二聚体、三聚体(trimer)、四聚体(tetramer)等。电泳是区分不同分子量蛋白质聚集体的常规手段,但由于Aβ寡聚体的亚稳态性,在电泳过程中会发生Aβ的自组装,从而影响实验结果。可通过光诱导蛋白交联(photoinduced cross-linking of unmodified proteins, PICUP)技术来稳定寡聚体的结构,以终止介尺度寡聚体动态组装过程[12]。通过PICUP并随后电泳后发现,不同类型的Aβ所组装形成的寡聚体各不相同:Aβ42能够形成五聚体(pentamer)、六聚体(hexamer),进一步会形成类似于原纤维的串珠结构;而Aβ40则会聚集成二聚体、三聚体、四聚体[12]。其中二聚体是毒性寡聚体的基本构件分子,三聚体可以组装形成分子量为56 kDa的Aβ*56或者Aβ十二聚体(dodecamer)。Aβ42十二聚体能够进一步组装形成线性的前原纤维(preprotofibrils),因此认为Aβ42十二聚体是Aβ42纤维形成的种子[15]。
寡聚体会通过减弱长时程增强过程(long-term potentiation, LTP)来影响记忆。LTP通过长时间强化神经信号来促进记忆的形成,因此LTP的减弱常与信号转导以及记忆的衰退有关。研究发现,来自于7PA2细胞的三聚体能够减弱LTP。同样,通过SEC分离AD患者大脑提取物得到的二聚体也能够衰减LTP,影响大鼠的记忆[7]。在AD转基因小鼠的脑内也发现了Aβ十二聚体,当把分离得到的十二聚体注射入大鼠的脑内后,通过水迷宫实验发现十二聚体也会导致记忆的下降。虽然二聚体、三聚体、十二聚体中哪种寡聚体的毒性最大有待进一步的探索,但目前一般认为高分子量(high molecular weight, HMW)寡聚体的毒性强于低分子量(low molecular weight, LMW)的寡聚体[16]。
2.2 按形貌、结构分类
介尺度寡聚体按形貌分类可分为球形、环形等。球形结构的Aβ寡聚体包括Aβ衍生扩散配体(Aβ-derived diffusible ligands, ADDLs)、淀粉样球体(amylospheroids, ASPDs)、球聚体(globulomers)等。
ADDLs是在AD患者的脑内检测到具有扩散性的球形寡聚体,粒径5~6 nm,分子量10~100 kDa,可以被ADDLs特异抗体NUx和纤维特异性抗体OC识别[17-18]。ADDLs不仅可以和成熟的神经元结合,产生神经毒性并诱导神经元自噬;还能减少神经元上的胰岛素受体并影响胰岛素的正常信号。此外,ADDLs能够诱导Tau蛋白过磷酸化并抑制LTP。
ASPDs是粒径10~15 nm、分子量158~669 kDa的完美球形结构[19]。由商业化Aβ在体外培养5 h形成。也存在于AD患者的脑内,是不参与Aβ纤维形成(off-fibril pathway)的寡聚体。ASPDs不与寡聚体抗体A11和纤维抗体OC产生反应,因此其构象与Aβ二聚体、Aβ十二聚体不同。HMW的ASPDs在神经退行的早期阶段通过影响Tau蛋白激酶Ⅰ/GSK-3β诱导细胞毒性[20]。近期的研究表明,蛋白酶体抑制会使兴奋性神经元中的ASPDs水平升高,导致ASPDs从轴突转移至树突。ASPDs会进一步分泌并结合至附近的Na+/K+-ATPase α3(NAKα3)神经元,引起NAKα3神经元的死亡[21]。
Globulomers是2005年发现的一种高度稳定的水溶性球形寡聚体,分子量60 kDa[22]。这种寡聚体可以由Aβ与SDS或脂肪酸培养得到,并在AD患者和转基因Tg2576小鼠脑内发现了此种寡聚体。与Aβ纤维不同,Globulomers同时含有平行和反平行的β-sheet结构。其通过减弱LTP,抑制AD患者突触的活跃度来影响认知[22]。
环形Aβ原纤维(annular protofibrils, APFs)具有环形或者孔形的结构,直径18~25 nm,可以稳定数月,并且能够被环形抗原纤维抗体检测[23]。北极突变型(E22G)Aβ40和野生型Aβ40均可以形成APFs。Aβ42原纤维寡聚体在亲水-疏水的界面的作用下也能够形成APFs。在脂质膜的环境中APFs能够形成β-桶(β-barrel)状结构。
尽管研究者对Aβ寡聚体的形貌有初步的了解,但由于Aβ寡聚体的多尺度性、暂态性和复杂性,原子尺度寡聚体结构的解析还存在较大的困难。通常需要计算机模拟和实验相结合的方法来确定特定寡聚体的结构[24]。图2列举了通过计算机模拟或者结构解析的手段得到的几种常见的寡聚体结构,包括:二聚体、五聚体、六聚体、Globulomers、β-barrel、环形孔(annular pore)等[25-27]。
图2
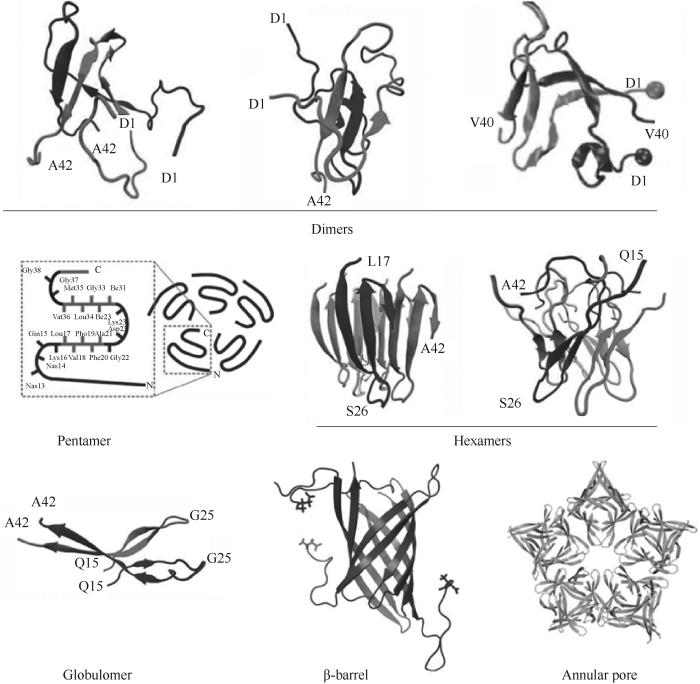
图2 Aβ寡聚体的结构[25-27]
Fig.2 Structures of Aβ oligomers[25-27]
2.3 按构象分类
不同介尺度寡聚体的构象各不相同,寡聚体的构象与其生物学效应最为相关,通常利用特异性抗体来识别区分不同生物功能的寡聚体。常见的抗体有A11、KW1、OC等。
A11抗体能特异性地与可溶性Aβ寡聚体相结合,不与Aβ单体或纤维产生作用[28]。A11也可与其他淀粉样蛋白所形成的寡聚体结合,如胰岛淀粉样蛋白(islet amyloid polypeptide, IAPP)、α-突触核蛋白(α‐synuclein)、溶菌酶等。因此,A11抗体能识别特定的寡聚体构象,而与氨基酸序列无关,此构象被认为是蛋白质聚集引起细胞毒性的关键、通用型的寡聚体构象。
KW1抗体能识别寡聚体中反平行β-sheet结构[29]。研究发现,KW1能够与Aβ寡聚体表面的疏水性区域(如Aβ18-20片段)产生反应。在人的大脑内也发现了KW1阳性的寡聚体,此种寡聚体会导致突触的功能性障碍。
OC抗体能够识别可溶性Aβ寡聚体和纤维。其中能与OC作用的寡聚体被称为纤维寡聚体(fibrillar oligomers),是因为这些寡聚体的构象与纤维类似,含有平行的β-sheet结构(in-register parallel β-sheet structure)。而与之相反,与A11作用的寡聚体为原纤维寡聚体(prefibrillar oligomers),其中含有反平行的β-sheet结构(out-of-register anti-parallel β-sheet structure)。由于原纤维寡聚体需要经过构象转变才能形成纤维寡聚体,因此纤维寡聚体的聚集速度比原纤维寡聚体快9倍[30]。
3 Aβ寡聚体的细胞损伤机制
虽然Aβ介尺度结构有待进一步解析,其动态组装机理尚不明确,但大量的研究表明可溶性Aβ寡聚体具有较高的神经毒性,其在AD的发病中起关键作用。由于Aβ寡聚体结构和性质以及体内环境的复杂性,寡聚体在细胞层面诱导毒性以及激活神经细胞死亡的机理有待进一步明确。目前普遍认为寡聚体通过以下三条途径对细胞产生损伤[31]:(1)寡聚体与细胞膜和膜受体产生相互作用,引起细胞膜的穿孔和细胞信号通路的改变;(2)寡聚体进入细胞后在细胞内积累,影响细胞器的功能和细胞内环境的稳态;(3)寡聚体在细胞间传播,导致淀粉样斑块的形成以及引发AD病理过程(图3)。
图3
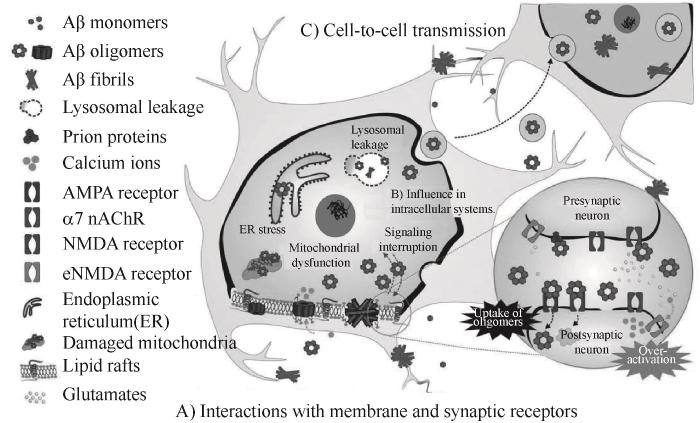
图3 Aβ寡聚体介导的细胞毒性[31]
Fig.3 Cytotoxicity triggered by Aβ oligomers[31]
3.1 Aβ寡聚体与细胞膜和膜受体的相互作用
细胞膜的完整性对于细胞活性的维持至关重要。研究表明,反平行β-sheet构象的Aβ寡聚体与孔蛋白结构相似,因此Aβ寡聚体能够引起细胞膜的穿孔。并且随着Aβ的纤维化,穿孔的细胞膜最终完全解体。在转基因Tg2576小鼠模型中发现,Aβ二聚体能够与细胞膜上的脂质筏(lipid rafts)产生相互作用并累积,形成局部较高的浓度,促进纤维化过程。由于脂质筏由胆固醇、GM1-神经节苷脂、鞘脂和突触受体组成,是神经信号的转导中心,因此Aβ寡聚体在脂质筏处的累积不仅会导致细胞的解体,还会干扰信号的转导,导致记忆下降。
此外,Aβ寡聚体能够与许多受体产生作用,调控下游信号通路,引起突触的功能性障碍以及神经退化。与Aβ能发生作用的受体有N-甲基-D-天冬氨酸受体(N‐methyl D‐aspartate receptors, NMDARs)、神经生长因子(nerve growth factor, NGF)受体、胰岛素受体(insulin receptors)、细胞内朊蛋白(cellular prion protein, PrPc)等。
3.2 Aβ寡聚体对细胞内过程的影响
胞外的Aβ可通过膜受体或神经元的内吞作用进入细胞内。此外,细胞器膜上APP产生的Aβ也可以直接在胞内累积。许多细胞器都是Aβ产生和聚集的位点,如线粒体、内质网、高尔基体、核内体、自噬体、溶酶体等。胞内Aβ寡聚体能够引起内质网压力的升高、Ca2+稳态失衡、线粒体损伤、蛋白质的水解以及细胞的凋亡。
Aβ寡聚体通过激活磷脂酶C的信号使内质网上Ca2+浓度失衡,导致Ca2+由内质网向细胞质释放,随之激活Caspase 3凋亡通路。Aβ寡聚体与线粒体膜作用,会引起细胞色素C的释放,启动凋亡过程。线粒体膜的解聚不仅代表线粒体功能的丧失,呼吸作用受到影响,还与突触功能异常以及记忆的损伤有关。当Aβ寡聚体与溶酶体作用时,会导致溶酶体膜的透化,增强溶酶体酶的外排,引起细胞凋亡。
3.3 Aβ寡聚体的传播
来自胞外或胞内的Aβ均可通过播种效应(seeding effect)在脑内扩散。Aβ寡聚体种子也可以通过细胞到细胞的传播机制进行扩散,受胞内体运输系统(endosomal trafficking system)的调控。此外,研究发现在不同Aβ亚型中,Aβ42传播性最强。寡聚体细胞到细胞的传播会导致细胞产生较大溶酶体囊泡,从而影响供体和受体细胞的溶酶体功能。
4 Aβ聚集的调控
由于Aβ寡聚体是Aβ聚集过程中毒性最高的组分,因此抑制Aβ纤维化、调控聚集过程中关键毒性介尺度结构的形成是减弱神经损伤最为有效、直接的途径。此外,也可通过抑制β-分泌酶的活性来减少Aβ单体的生成,利用胰岛素降解酶、脑啡肽酶等清除Aβ单体,或重塑Aβ聚集体等方式来调控Aβ聚集所产生的毒性,以达到治疗AD的目的。由于Aβ介尺度结构和聚集过程的复杂性,目前的调控/抑制剂虽然能够减弱细胞毒性,对于其作用和调控机理的研究还有待进一步深入,以下就常见的调控/抑制剂包括小分子类、多肽类、蛋白类和纳米类调控/抑制剂等进行介绍和归纳总结[32]。
4.1 小分子类抑制剂
天然小分子抑制剂具有分子量小、Aβ亲和性高等优点。迄今为止已经鉴定了两类小分子抑制剂:多酚类和非多酚类小分子。多酚类小分子含有一个或多个酚羟基,根据“π-stacking”理论,多酚类化合物的苯环可能竞争性地与Aβ的芳香基团发生作用,类似于三明治式地夹杂在两个氨基酸苯环残基之间,阻止Aβ单体间π-π相互作用。此外,多酚类化合物可能通过疏水性相互作用抑制淀粉样纤维的生长。常见的多酚类抑制有:茶多酚((-)-Epigallocatechin-3-gallate, EGCG)、白藜芦醇(Resveratrol)、姜黄素(Curcumin)、巴西木素(Brazilin)、苏木精(Hematoxylin)等(图4)。非多酚类小分子包括生物碱、黄酮类、苷类、吩嗪等。
图4
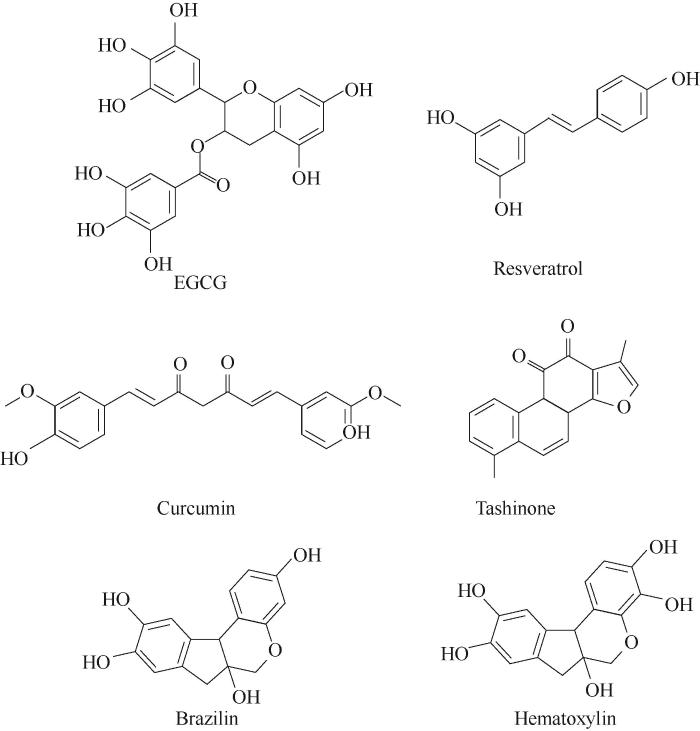
图4 调控Aβ聚集的小分子抑制剂
Fig.4 Small molecules that show the modulation capability of Aβ aggregation
来自于绿茶提取物的EGCG是研究最为广泛的小分子抑制剂,已进入临床Ⅲ期试验。EGCG不仅能抑制Aβ的纤维化,还能够解聚成熟的Aβ聚集体,使之形成大量无定形、非毒性的Aβ寡聚体[33-34]。当金属离子(如Cu2+、Zn2+等)存在时,EGCG又能与金属离子、Aβ形成三元复合物,抑制金属离子诱导的Aβ聚集,有效缓解Aβ-金属所引起的细胞毒性[35]。通过等温滴定量热(isothermal titration calorimetry, ITC)实验测定EGCG与不同Aβ片段结合的热力学参数后发现,疏水相互作用和氢键结合发生于Aβ的不同区域。氢键结合主要发生于Aβ1-16片段,而疏水相互作用主要作用于Aβ17-42片段[36]。此外,分子动力学模拟结果表明,EGCG能够将水分子从Aβ的表面排开,并直接与Aβ产生作用[37]。研究显示,非极性作用对EGCG-Aβ结合能的贡献大于71%,而极性作用对其结合的贡献较小。Aβ中有12个氨基酸残基均能够与EGCG产生作用,包括Phe4、Arg5、Phe19、Phe20、Glu22、Lys28、Gly29、Leu34~Gly37、Ile41。非极性相互作用主要由疏水性氨基酸残基(Phe、Met、Ile)的侧链和一些非疏水性氨基酸残基(Lys28、Gly29)的主链提供,而极性相互作用主要由Aβ的主链(Gly29、Gly37)提供。Ahmed等[38]进一步利用NMR技术对EGCG重塑Aβ寡聚体的分子机理进行了探究。发现EGCG对Aβ组装体的调控符合Hill-Scatchard模型。当与EGCG结合后,Aβ寡聚体溶剂暴露程度降低。此过程扰乱了Aβ单体与原纤维的接触,从而阻断了表面催化的二次成核过程,降低了毒性聚集体的生成。
从中草药丹参中提取得到的丹参酮(Tashinone),已广泛应用于治疗心血管、心肌梗死、中风等疾病。研究发现,丹参酮具有显著的神经保护作用;能够有效抑制Aβ聚集,解聚成熟的Aβ纤维,并减少Aβ聚集诱导的细胞毒性。对于Ⅱ型糖尿病相关的人胰岛淀粉样多肽(human islet amyloid polypeptide, hIAPP)的聚集,丹参酮也具有较强的抑制作用[39]。
巴西木素(Brazilin)是来源于苏木(Caesalpinia sappan)的一种天然多酚类化合物,具有Aβ聚集抑制和纤维解聚的双重性功能。在低浓度下,巴西木素的抑制能力强于EGCG。研究表明,巴西木素能够将Aβ单体和纤维转变为无定形的聚集体,阻止一次成核和纤维催化的二次成核过程。分子模拟结果显示,巴西木素能通过疏水和氢键作用与Aβ组分结合,抑制其纤维化;并通过氢键作用干扰D23~K28分子间盐桥的形成,进而重塑Aβ纤维[40]。此外,巴西木素还能够抑制来自于前列腺酸性磷酸酶39肽(prostatic acidic phosphatase, PAP248-286)的聚集,有效降低被HIV感染的风险[41]。与巴西木素的结构类似,另一种来自于苏木的天然小分子化合物苏木精(Hematoxylin),也是有效的Aβ聚集抑制剂[42]。苏木精能减少聚集过程中β-sheet结构的形成,诱导Aβ形成反平行结构,改变聚集路径,显著缓解由Aβ聚集所致的细胞毒性。
小分子类抑制剂虽然能在体外有效抑制Aβ的聚集、解聚Aβ纤维,但小分子类抑制剂的专一性差、体内循环时间短、稳定性不足、易被降解和代谢,上述缺点限制了小分子抑制剂的体内应用。因此,需要设计适宜的药物递送体系,以提高小分子的生物利用度。
4.2 多肽类抑制剂
多肽类抑制剂具有易于合成修饰、生物相容性好、特异性高、分子量小、无免疫原性、理化性质简单、可通过理性设计得到等优势[43],因此近年来多肽药物发展迅速。目前,已有60多种多肽药物上市,近140种正在进行临床试验,至少500种处于临床前评估阶段。Aβ多肽类抑制剂通常来自于Aβ自身序列,或根据特定原则构建的多肽库,随后利用分子对接或者分子动力学模拟的方法进行筛选,并通过试验的手段验证得到。此外,还可以对先导肽进行衍生、改造,以得到抑制能力更强、功能性更多的多肽抑制剂(表1)。
表1 基于Aβ核心疏水区设计的多肽抑制剂总结
Table 1
| 多肽序列 | 评论 | 文献 |
|---|---|---|
| KLVFF | 基于Aβ核心疏水区设计的多肽抑制剂 | [44] |
| LPFFD | 抑制大鼠体内淀粉样斑块的生成和沉积 | [45] |
| LVFFARK | 通过增加带电氨基酸以增强抑制剂与Aβ的结合 | [46] |
| c-LVFFARK | 通过首尾环化的方式降低自聚,增强与Aβ的亲和性 | [47] |
| LVFFARKHH | C-末端增加HH,引入Cu2+螯合功能 | [48] |
| Car-LVFFARK | N-端引入Car,能够抑制Cu2+介导的Aβ聚集和ROS | [49] |
| L-RTHLVFFARK | N-端增加RTH三肽,能够选择性地螯合Cu2+ | [50] |
| D-RTHLVFFARK | 多功能抑制剂,通过手性转变提高了抑制和重塑能力 | [51] |
新窗口打开| 下载CSV
基于Aβ序列设计的抑制剂,根据多肽序列的来源或作用区域可分为Aβ核心疏水区和C-端结构域。Aβ核心疏水区在Aβ聚集中发挥着关键作用,被称为“自识别序列”。由于多肽的纤维化是Aβ单体不断的自我识别过程,因此来自于Aβ核心疏水区的序列能够与Aβ分子中相似的片段结合,从而抑制聚集。为评估Aβ核心疏水片段KLVFF中各氨基酸在抑制Aβ聚集中的作用,Tjernberg等[44]用Ala分别取代了KLVFF序列中的Lys16、Leu17、Phe20,以此设计了短肽抑制剂KLVFF。Soto等[45]发现,倾向于形成β片层构象的序列能够促进Aβ的纤维化过程。因此,其将Leu17替换为具有阻断β片层生成作用的Pro,设计出了β-折叠破坏肽(β-sheet breakers)LPFFD。LPFFD不仅能够抑制Aβ的聚集,还能够解聚成熟的Aβ纤维,阻止神经细胞的死亡,并且也能抑制体内淀粉样斑块的形成和沉积。
虽然KLVFF或LVFFA均能通过疏水和氢键作用与Aβ结合,但其抑制Aβ纤维化的能力较弱,需要经过更进一步的设计和改造来提高抑制能力。通过分析Aβ的疏水核心区以及附近的氨基酸发现,疏水核心之后的是带负电的氨基酸残基E、D。因此如果在多肽抑制剂LVFFA的末端增加带正电的氨基酸,将会通过静电相互作用来加强抑制剂与Aβ的结合。为此引入了带正电的R、K,设计了七肽抑制剂LVFFARK(LK7)[46]。当LK7与Aβ浓度相同时,能够抑制68%的ThT荧光强度,此时抑制效果最好。但由于自身的聚集性质,进一步增大LK7的浓度反而会导致抑制效果的降低以及明显的细胞毒性。为提高LK7的抑制能力、解决LK7自聚的问题,Ma等[47]通过首尾环化的方式设计了环化LK7(cLK7)。与线性的LK7相比,cLK7具有较高的酶解稳定性、较低的细胞毒性。cLK7与Aβ较高的亲和性有利于其结合到Aβ单体上,稳定Aβ的构象,从而显著抑制Aβ的纤维化和细胞毒性。
由于AD病因的复杂性,单纯的Aβ抑制剂往往很难治愈AD。AD的其他病理学特征如过渡态金属离子(Cu2+、Zn2+)紊乱,能够促进Aβ的聚集,形成毒性更高的聚集体[52-53]。此外,过渡态金属离子会催化类芬顿反应(Fenton-like reaction),生成大量的活性氧自由基(reactive oxygen species, ROS),对细胞产生氧化损伤[54]。因此,开发具有金属螯合能力、Aβ抑制能力的多肽具有重要意义。双/多功能抑制剂不仅能够抑制Aβ自身的聚集,还能够抑制金属离子介导的聚集和产生的ROS,以及解聚Aβ-金属复合物。在构建双/多功能抑制剂方面也有广泛的研究。如以LK7为基础,在其C-末端引入具有Cu2+螯合能力的HH,可构建双功能抑制剂LVFFARKHH[48];在其N-端分别引入肌肽(carnosine, Car)、三肽RTH,可构建Car-LK7[49]、RTHLVFFARK(L-RK10)[50]等。上述多功能多肽抑制剂不仅具有金属螯合能力,更重要的是由于亲水性基团的引入,降低了LK7的自聚趋势,提高了其抑制性能。
Aβ具有特定的空间构象,对抑制剂存在明显的手性选择和偏好性。因而多肽抑制剂的手性转变是提高抑制效率的有效方式。Liu等[51]对多功能抑制剂L-RK10进行了手性转变,以此设计了D-RK10。与L型相比,D-RK10不仅具有更高的抑制能力,还具有类似的ROS抑制、抗氧化损伤性质;此外D-RK10能够重塑Aβ-Cu2+聚集体,阻止解聚碎片的重聚。
通过环化、手性转化、引入非天然氨基酸、N-甲基化等方式对多肽抑制剂进行衍生,能够提高抑制剂的抑制能力、增加其功能性、增强抗蛋白水解能力以及提高BBB渗透性。虽然多肽类抑制剂具有较高的生物相容性和Aβ特异性,但是多肽类抑制剂还存在如下缺点有待克服,如:多肽类抑制剂普遍疏水性较强、易于自聚,由此引起抑制能力的降低以及细胞毒性的升高;多肽类抑制剂抑制效果差、有效作用浓度偏高、体内稳定性不足、易酶解、半衰期短,因此还需要进一步的优化和改进。
4.3 蛋白类抑制剂
存在于人体内的天然蛋白质可作为分子伴侣,调控Aβ的聚集和神经毒性。已经发现大量蛋白质如:人血清白蛋白(human serum albumin, HSA)、热休克蛋白(heat shock protein, HSP)、丛生蛋白、甲状腺素转运蛋白(transthyretin, TTR)、朊蛋白PrPc、溶菌酶(lysozyme, Lys)等均能够与Aβ产生相互作用。蛋白类抑制剂具有较高的生物相容性、易于修饰的优点。
血清白蛋白是血清中含量最丰富的蛋白质,通常作为营养分子、代谢物、金属离子、药物和其他分子的转运蛋白。根据来源,可分为HSA和牛血清白蛋白(bovine serum albumin, BSA)。HSA和BSA的结构较为相似,有76%的序列相似性。美国FDA已批准BSA用于药物制剂的生产和应用。HSA可以通过疏水口袋以1∶1比例与Aβ结合。此外,HSA含有两个金属结合位点,能够螯合Cu2+、Zn2+等金属离子,有效抑制金属离子诱导的Aβ聚集。为提高血清白蛋白的抑制能力,Xie等[55]通过引入负电荷的方法合成了酸化的白蛋白(acidulated BSA, A-BSA)。在与Aβ等摩尔浓度培养时,A-BSA和BSA能分别减少47%和25%的ThT荧光强度。A-BSA能够更显著地抑制Aβ纤维化,有效缓解Aβ聚集介导的细胞毒性(图5)。基于此,研究者提出了疏水结合-静电排斥(hydrophobic binding-electrostatic repulsion, HyBER)的机理假说。此假说认为Aβ通过疏水作用结合到A-BSA表面;同时由于Aβ和A-BSA带同种电荷,对结合到A-BSA表面的Aβ起到静电排斥作用。上述两种相反的作用使Aβ维持伸展的结构,而非有利于聚集的β-sheet结构。因此A-BSA能够调控Aβ的聚集并改变其细胞毒性。由于白蛋白固有的金属螯合能力,A-BSA、A-HSA也可以用于调控Cu2+、Zn2+介导的Aβ聚集和毒性[56-57]。此外,对HSA修饰金属螯合剂亚氨基二乙酸(iminodiacetic acid, IDA),可进一步提高HSA的金属螯合容量,有效抑制高浓度金属离子所产生的ROS,以及金属离子诱导的Aβ聚集[58]。
图5
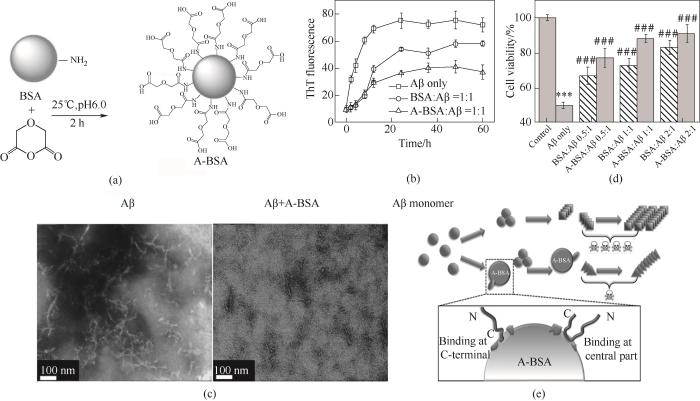
图5 A-BSA抑制Aβ聚集[55]
(a) A-BSA合成示意图;(b) Aβ聚集动力学实验;(c) Aβ在无或有A-BSA条件下培养24 h的TEM图片;(d) MTT细胞活性实验;(e) HyBER假说示意图
Fig.5 A-BSA inhibits Aβ aggregation[55]
(a) Schematic illustration of the preparation of A-BSA; (b) Aggregation kinetics of Aβ incubated with BSA/A-BSA; (c) TEM images of Aβ incubated (A) without or (B) with A-BSA for 24 h; (d) MTT cell viability of PC-12 cells treated with Aβ and different BSA/A-BSA concentrations; (e) Schematic representation of the HyBER hypothesis
以血清白蛋白为基础,通过增加表面正电荷也可以增强其抑制效果。Wang等[59]利用BSA表面偶联乙二胺的方法制备了碱化的BSA (BSA-B)。研究发现,在较低浓度条件下,BSA-B表现出高效的抑制能力。碱化修饰增强了BSA与Aβ之间的静电相互作用,实现了BSA与Aβ结合方式由单位点的疏水结合向多位点的静电结合转变,从而能够与不同的Aβ组分产生相互作用,有效抑制Aβ的纤维化过程。对HSA进行碱化修饰也得到了类似的结果。在碱化HSA (HSA-B)基础上进一步偶联Aβ特异性近红外荧光探针、BBB穿膜肽可制备新型的诊疗试剂(HSA-BFP)[60]。HSA-BFP不仅能抑制AD线虫体内Aβ的沉积、缓解由聚集引起的瘫痪,还能实现体内Aβ斑块的荧光检测,是一种多功能Aβ诊疗剂。
其他的蛋白质类抑制剂,也可通过表面化学改性的方式提高抑制能力,或赋予其新的功能。如:对溶菌酶进行碱化修饰会增强其抑制能力[61];偶联IDA,引入了Zn2+螯合性质[62];修饰多功能多肽RK10,实现了对Cu2+介导Aβ聚集的抑制,以及对Cu2+-Aβ聚集体的重塑等[63]。
4.4 纳米材料类抑制剂
随着纳米技术的迅速发展,纳米材料的生物医学应用受到了广泛的关注。纳米材料具有粒径小、比表面积较大、易于修饰和改性的特点[64-66]。各类纳米材料,包括聚合物纳米粒子、金属纳米粒子、2D纳米材料、碳纳米材料以及功能性纳米粒子在抑制Aβ聚集和治疗AD方面均有涉及(图6)。可根据特定的生理微环境或药物作用机制来理性设计不同的纳米材料,用于提高药物的递送效率、实现疾病的治疗。
图6
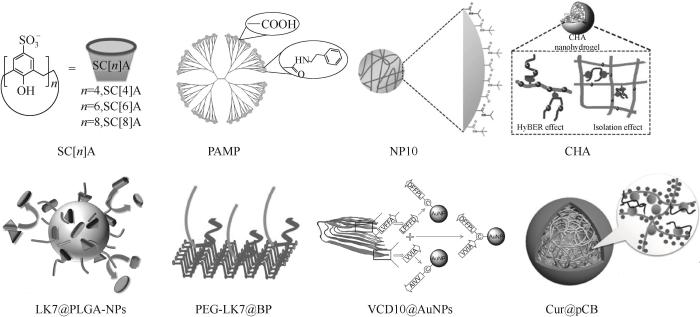
图6 纳米材料用于调控Aβ聚集[46,67-73]
Fig.6 An overview of nanomaterials for modulation of Aβ aggregation[46,67-73]
超分子化合物可用于抑制Aβ聚集。第二代大环超分子如β-环糊精(β-cyclodextrins, β-CD)、葫芦脲等能够通过疏水腔调控Aβ的聚集[74]。此外,作为一种优良的药物载体,将疏水性的多肽类抑制剂LK7偶联于β-CD上,能显著提高LK7的溶解性,有效增强抑制效率[75]。以杯芳烃为代表的第三代超分子主体化合物不仅继承了前两代的特点,还具有易衍生的优势。磺化杯芳烃(para-sulfonato-calix[n]arene, SC[n]A)是杯芳烃的一种衍生物,可以通过主客体化学与多种氨基酸产生静电、疏水等相互作用,具备抑制Aβ聚集的潜质。Wang等[67]研究了SC[n]A(n=4, 6, 8)对Aβ聚集的影响,发现抑制作用的强弱为:SC[8]A>SC[6]A>SC[4]A。由于SC[8]A的分子环最大,其柔性也最大,有利于与Aβ发生相互作用,因此抑制效果最好。
树枝状聚合物是高度支化的高分子纳米材料,包括聚酰胺树枝状聚合物(polyamidoamine dendrimer, PAMAM)、聚乙烯亚氨树枝状聚合物、聚丙烯亚胺树枝状聚合物等。在药物递送、可控释放、诊断治疗等方面均有应用。为验证抑制剂通过HyBER机理调控Aβ聚集的正确性,研究者在5代(G5)羧基化PAMAM的表面偶联了疏水性的苯乙胺(phenethylamine, PEA),合成了带有负电的疏水性抑制剂(PAMP)[68],发现适宜取代度的PAMP具有最佳的抑制效果,能够通过疏水结合-静电排斥作用来调控聚集并缓解细胞毒性。此外,通过研究不同代数PEA修饰的PAMAM对Aβ聚集的抑制后发现,高代数的G5-P、G6-P具有抑制能力,而低代数的G3-P、G4-P无显著的抑制作用。因此,疏水基团和负电荷基团需要在空间上满足一定的范围才能发挥HyBER作用。当超过此范围后,两种相反的作用力不能使Aβ维持伸展的构象,因而不具有抑制能力[76]。HyBER假说中疏水与静电作用对抑制效率的影响还可以通过其他聚合物纳米粒子进行证明和进一步的探究,如:N-叔丁基丙烯酰胺与丙烯酸通过共聚得到的聚合物纳米粒子NP10[69]、姜黄素修饰的透明质酸纳米粒子(curcumin-hyaluronic acid conjugate, CHA)等[70]。
以纳米粒子为载体,将多肽或小分子类抑制剂偶联于纳米粒子上,可以改善多肽或小分子的水溶性,避免其由于自聚而导致的抑制能力降低以及细胞毒性的升高,增强抑制效果。如将多肽抑制剂LK7偶联于粒径为150 nm的聚乳酸-乙醇酸[poly(lactic-co-glycolic acid), PLGA]纳米粒子上,制备多肽功能化的纳米粒子LK7@PLGA NPs,有效克服了LK7高浓度条件下自聚的缺点[46]。将LK7负载于新型的2D纳米材料黑磷(black phosphorus, BP)片层表面,并修饰聚乙二醇PEG,合成了PEG-LK7稳定的BP (PEG-LK7@BP)[71]。在有效防止BP降解的同时,较大的BP片层表面为LK7提供了附着平台,提高了LK7的局部浓度,有利于LK7通过“多价”作用与Aβ组分结合,增强了LK7对Aβ纤维化的抑制。再比如,将所设计的嵌合肽VVIA-CLPFFD(VCD10)偶联于金纳米粒子(AuNPs)上,合成VCD10偶联的AuNPs (VCD10@AuNPs)。多肽在AuNPs表面上特定的趋向和构象,能够促进其与Aβ产生相互作用,显著提升多肽的抑制能力[72]。在0.1 nmol·L-1的浓度下,即可将细胞活性由48%提升至82%。
作为一类亲水性聚合物,两性离子聚合物由于其优异的生物相容性而受到重视。常见的两性离子有聚羧基甜菜碱[poly(carboxybetaine), pCB]、聚磺酸基甜菜碱[poly(sulfobetaine), pSB]等。为改善小分子或多肽抑制剂的药效,可将其偶联于两性离子聚合物上。Zhao等[73]将姜黄素(Curcumin)修饰于pCB上,合成了姜黄素偶联的pCB纳米粒子(Cur@pCB)。Cur@pCB自组装形成粒径为120~190 nm的纳米凝胶。其表面的水化层能稳定结合在姜黄素上的Aβ,阻止Aβ构象向β-sheet结构的转变。与游离的姜黄素相比,同浓度的Cur@pCB抑制能力明显提升。类似地,也可将LK7偶联于pCB上,制备多肽修饰的聚合物纳米粒子LK7@pCB[77]。通常情况下,抑制剂通过抑制β-sheet结构的生成来降低聚集所诱导的细胞毒性。然而Wang等[78]发现,将LK7偶联于带有二甲基侧链的两性离子pID(LK7@pID)后,促进了Aβ的聚集,此过程扰乱了正常的Aβ纤维化,生成了大量异质的纤维聚集体,因此降低了细胞毒性。
此外,一些碳材料,如石墨烯等可用于检测Aβ单体、抑制Aβ的聚集。Gao等[79]通过水热法合成了粒径为5 nm的超小氮掺杂碳化聚合物点(nitrogen-doped carbonized polymer dots, CPDs)。CPDs不仅能通过静电吸引、氢键结合以及疏水相互作用抑制Aβ的纤维化,减少聚集所产生的毒性。还能在数秒至数分钟的时间尺度内快速解聚Aβ纤维。更重要的是,当与Aβ结合后,CPDs表现出红色荧光增强的性质。因此CPDs可用于体内Aβ斑块的成像。体内实验结果表明,CPDs能够通过清除Aβ斑块,延长转基因线虫CL2006的生存寿命,是一种多功能的诊疗试剂(图7)。
图7
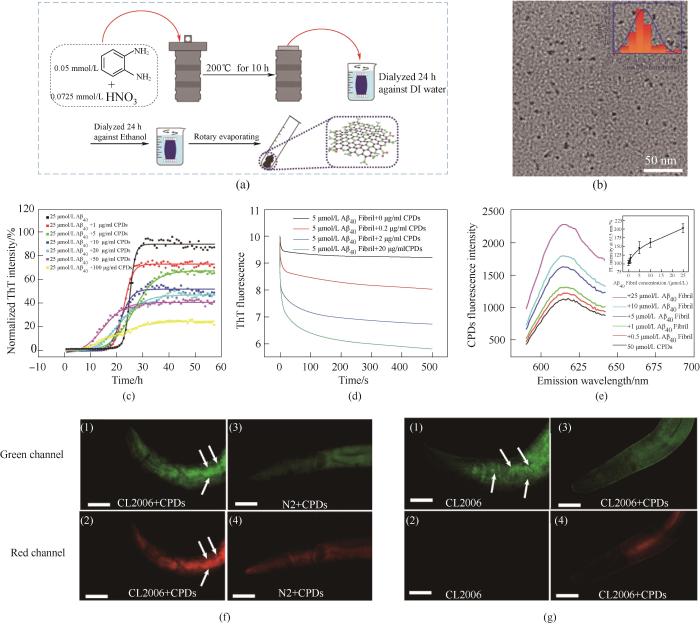
图7 CPDs靶向Aβ聚集的多功能性[79]
(a) CPDs的合成示意图; (b) CPDs的TEM图片; (c) 通过ThT荧光实验测定CPDs抑制Aβ聚集; (d) CPDs快速解聚Aβ纤维; (e) CPDs荧光检测Aβ纤维; (f) CPDs检测线虫体内Aβ斑块; (g) CPDs抑制线虫体内Aβ沉积
Fig.7 The multifunctionality of CPDs targeting Aβ aggregation[79]
(a) Schematic representation of the synthesis of CPDs; (b) TEM image of CPDs; (c) CPDs inhibit Aβ aggregation measured by ThT fluorescence assay; (d) Fast disaggregation of Aβ fibrils by CPDs using a stopped-flow spectrometer; (e) Detection of Aβ fibrils by the fluorescence of CPDs; (f) Imaging Aβ plaques in CL2006 nematodes; (g) CPDs suppress Aβ accumulation in nematodes
除常规纳米材料外,光、超声等外场响应的功能性纳米材料也被用于调控Aβ的聚集,如光热纳米材料、光氧化纳米材料等[80]。纳米马达是一类能够受外场激发、在微观尺度自主运动的纳米粒子,已应用于强化化学反应、体内药物的靶向递送、疾病治疗等诸多领域。研究者将多肽抑制剂D-RK10负载于近红外(near infrared region, NIR)光驱动的Janus纳米马达上,构建了抑制剂修饰的纳米马达(JNM-I)[81]。在NIR光的照射下,JNM-I通过“自热泳”力产生显著的运动。NIR光驱动的JNM-I能够增强抑制剂与Aβ组分之间的相互碰撞,显著提高抑制剂的抑制能力,减轻Aβ聚集导致的毒性,阻止Aβ构象由无规则卷曲向β-sheet的转变。此外,由于JNM-I的主动运动特性和光热作用,NIR光也能增强其BBB透过性,这有利于AD药物的递送和进一步体内应用(图8)。此研究以纳米马达为载体实现了对Aβ聚集过程的调控与强化,创造性地将过程强化理论应用于与Aβ聚集相关的介尺度科学的研究。
图8
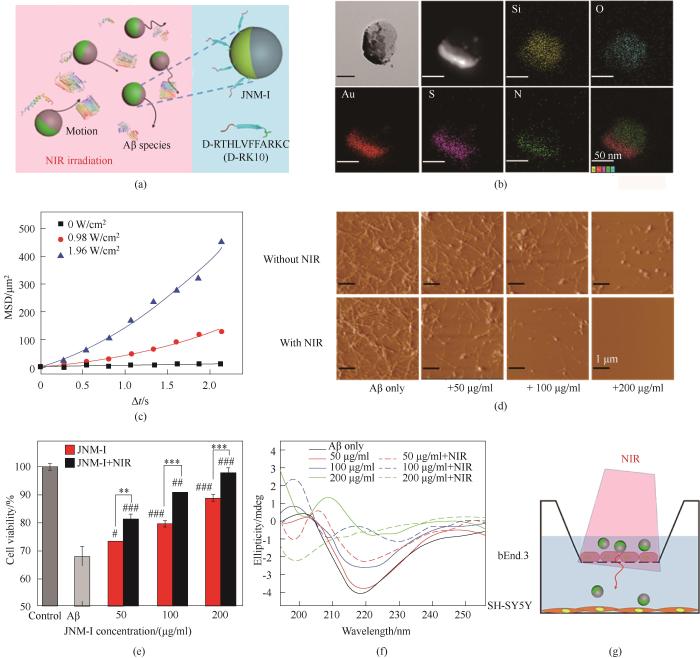
图8 NIR光驱动的JNM-I用于强化抑制Aβ纤维化[81]
(a) JNM-I通过运动作用增强抑制剂与Aβ组分之间的作用; (b) JNM-I的TEM和能谱图片; (c) 不同激光强度下MSD随时间间隔变化的曲线; (d) Aβ聚集体的AFM图片; (e) 在有或无NIR光照条件下SH-SY5Y细胞与JNM-I培养的MTT细胞活性实验; (f) Aβ聚集48 h后的CD光谱; (g) NIR驱动的JNM-I穿透BBB
Fig.8 NIR light-powered JNM-I for intensified inhibition of Aβ fibrillogenesis[81]
(a) Enhancing the interactions between inhibitors and Aβ species by the active motion of JNM-I; (b) TEM image and EDS mapping of JNM-I; (c) Curves of MSD vs time interval at varied laser densities; (d) AFM images of Aβ aggregates; (e) MTT cell viability of SH-SY5Y cell incubated with JNM-I and treated with or without NIR light; (f) CD spectra of Aβ after incubation for 48 h; (g) NIR light-propelled JNM-I to cross BBB
5 结论与展望
随着人口老龄化的日益加剧,AD已成为医学、健康领域重点关注和亟待解决的问题之一。目前临床上缓解AD认知障碍的药物主要有4种,包括3种胆碱酯酶抑制剂(Donepezil, Galantamine, Rivastigmine)和1种N-甲基-D-天冬氨酸受体拮抗剂(Memantine)。经过多年的研究,以Aβ级联假说为基础的药物设计已取得了一定的结果,并开发出了一系列的Aβ抑制剂,有些已经获得FDA的批准或进入临床实验阶段。如Biogen公司开发的单抗类药物Aducanumab在2021年6月获得了美国FDA的加速审批,成为第一个抗Aβ聚集的药物[82]。但目前针对Aβ的研究还存在如下缺点,需要在今后的研究中得以解决。
(1)由于AD病因的复杂性,Aβ的聚集只是其中一种因素。其他方面,如过渡态金属离子紊乱、Tau蛋白过磷酸化、神经炎症等均是AD的重要病理学特征,在这些方面还应该进行更为广泛、充分的研究,以针对AD病因设计更为可行的候选药物。例如:肠道菌群紊乱所诱发的神经炎症是AD的重要发病机制,调控肠道菌群可改变氨基酸衍生物的代谢进而影响免疫细胞的浸润。为此中国科学家研制了首个靶向脑-肠轴的寡糖类AD药物GV-971,并于2019年11月获得国家药品监督管理局的批准在国内上市。
(2)对于Aβ聚集的关键介尺度结构、介尺度动态组装机理以及Aβ聚集的生物学作用的理解和认识还远远不足。因此需要通过更为综合、先进创新的手段解析Aβ寡聚体的基本结构,揭示聚集过程的分子机理,并阐释其引起细胞损伤的机制。
(3)目前的Aβ抑制剂虽然能阻止Aβ的聚集,但其具体的作用原理还有待更深入的探究,需要针对特定的介尺度结构设计更为专一、高效的抑制剂,实现对聚集的调控与抑制。此外,抑制剂的BBB透过性、体内药效的评估等方面还应该进行广泛的研究。
(4)在实际应用中,单纯的Aβ抑制剂可能不能取得较好的临床结果。因此,需要在Aβ抑制剂的基础上对其进行设计改造,如:诊疗一体化的设计、多功能设计、可控释放设计等,以满足临床的需要。

- 刚刚!2026年中科院分区,公布!本次看点:中科院分区变更为新锐分区;不再单独发布预警期刊;37种期刊“under review”~
- 这些重要报纸理论版都支持邮箱投稿!回复极快!
- GB/T 7714-2025与GB/T 7714-2015相比,变更了哪些,对期刊参考文献格式有什么影响?
- 别被这个老掉牙的报纸理论版投稿邮箱误导了!最新核实91个报纸理论版投稿邮箱通道,一次集齐
- 喜报!《中国博物馆》入选CSSCI扩展版来源期刊(最新CSSCI南大核心期刊目录2025-2026版)!新入选!
- 2025年中科院分区表已公布!Scientific Reports降至三区
- 国内核心期刊分级情况概览及说明!本篇适用人群:需要发南核、北核、CSCD、科核、AMI、SCD、RCCSE期刊的学者
- CSSCI官方早就公布了最新南核目录,有心的人已经拿到并且投入使用!附南核目录新增期刊!
- 北大核心期刊目录换届,我们应该熟知的10个知识点。
- 注意,最新期刊论文格式标准已发布,论文写作规则发生重大变化!文字版GB/T 7713.2—2022 学术论文编写规则

