 0373-5939925
0373-5939925 2851259250@qq.com
2851259250@qq.com

高分子复合材料中键合剂在不同纳米填料表面的吸附能计算
由高分子、填料、键合剂及其他各种功能助剂组成的高分子复合材料具有耐腐蚀性、成本低、适用性强、制备工艺简单、耐疲劳性以及减震性能强等特点[1]。这些优异的特性使得高分子复合材料在含能材料、轮胎、航空航天、建筑等方面有着广泛的应用[2]。填料和键合剂是高分子复合材料的重要组分,键合剂与填料表面的键合能力显著影响高分子复合材料的抗拉伸、黏弹性等性能。
轮胎是一种常见的高分子复合材料。发展高耐磨损、高抗湿滑、低滚阻的新一代轮胎是轮胎工业的重要挑战。在轮胎的生产用料中,填料的用量仅次于聚合物(主料),因此,填料也常被称作副料。填料的加入能提高聚合物复合材料的性能,改善轮胎的抗湿滑性、耐疲劳性以及耐低温耐高温能力等[3]。炭黑和二氧化硅是常用的轮胎填料。其中,炭黑为层状结构,化学性能稳定且无毒,加入之后可以使轮胎有较高的耐疲劳性,增强轮胎的力学性能,提高轮胎耐磨性的同时可降低轮胎的滚动阻力[4]。二氧化硅表面有很强的吸附特性,比表面积大,可以实现对有机分子的多层吸附,提高轮胎的抗撕裂性能[5]。调节炭黑和二氧化硅的添加量、颗粒大小和级配可生产出满足不同需求的轮胎。此外,应用于航空航天领域的含能高分子复合材料中固体填料含量更多,其质量分数达50%~70%,如高氯酸铵颗粒是一种常见的强氧化剂,作为填料加入推进剂后可在燃烧过程中快速分解提供大量的氧气,使推进剂能够充分燃烧提供强劲推力[6]。
在轮胎和推进剂体系中,固体填料与聚合物之间的相容性都是影响高分子复合材料的关键因素。当聚合物主体和填料发生“脱湿”时,即聚合物主体与固体填料发生界面剥离,高分子复合材料的力学性能、安全性都显著下降。键合剂填充在聚合物主体和固体填料形成的界面,一端与固体填料表面键合,一端与聚合物分子键合,可使聚合物主体与固体填料紧密结合在一起,从而能较大程度防止“脱湿”现象的发生。显然,键合剂在填料表面的吸附能力非常重要,其吸附能力强弱是筛选、开发合适键合剂或填料的重要依据。因此,开展键合剂与固体表面相互作用计算具有重要的应有价值[7]。此外,该吸附机理研究还广泛应用在电子领域[8-10]、生物传感器[11]、环保吸附剂[12]等相关器件和功能材料开发上。
键合剂分子在填料表面的吸附能大小难以用传统方法进行测定表征。近年来,研究人员发展了采用第一性原理计算的方法来研究小分子在固体表面的吸附。张志强等[13]利用第一性原理研究了二氧化硅吸附乙醇分子,发现二氧化硅表面接枝羟基后,易与乙醇分子中的氧原子形成氢键,从而增强二氧化硅对乙醇的吸附作用;Irfan等[14]基于密度泛函理论计算,研究了有机分子在不同层数炭黑材料上的吸附,发现双层炭黑要比单层炭黑具有更大的吸附能;Gao等[15]采用密度泛函理论研究了炭黑吸附尾气中的NO,通过研究NO在炭黑不同位置上的吸附能并开展态密度分析,发现该吸附过程为物理吸附,并确定了最佳吸附位点;Li等[16]研究了多芳环有机物与炭黑的相互作用,发现炭黑对有机分子的吸附具有更快、更灵敏的特性,从而在去除持久性芳香族化合物的污染方面有很大的潜力。
前期研究表明,基底材料的层数[17]影响其与键合剂分子的相互作用,而且基底表面的缺陷和形貌也会改变分子的吸附特性[17-20]。高氯酸铵、炭黑和二氧化硅对键合剂分子的吸附也得到了较多关注[13-14,21],但材料厚度、表面改性等因素对键合剂分子吸附的影响尚未见报道。基于此,本文通过研究T313和TEA分子在不同厚度高氯酸铵表面的吸附,分析DPG、DPTU和DOTG三种键合剂分子在不同层数炭黑及二氧化硅表面的吸附差异,探索炭黑的缺陷位点(单空位、双空位和Stone-Wales)及二氧化硅表面接枝羟基对吸附的影响,阐明不同键合剂分子在多种界面的吸附机理。
1 计算模型和方法
利用Vienna Ab-initio Simulation Package (VASP)软件[22]计算吸附能,采用GGA- PBE[23]泛函,电子波函数采用平面波基组展开,多电子体系之间的相互作用采用超软贋势[24],截断能设置为400 eV。将三种填料的原始晶胞切割,二氧化硅和高氯酸铵选用(010)晶面,炭黑选用(001)晶面,而后建立6×3的高氯酸铵超胞、6×6的炭黑超胞和6×2的二氧化硅超胞,超胞上方添加20 Å(1 Å=10-10 m)真空层[25]以消除周期性边界条件对计算结果的影响,三种填料的原始晶胞见图1。建立五种键合剂(TEA、T313、DPG、DPTU和DOTG)的分子模型,其分子结构、静电势分布如图2所示,分子表面静电势分布由Multiwfn[26-27]和VMD[28]共同绘制。从图中可看出,极性原子如S、F、N、O静电势为负所以偏蓝色,而与极性原子相连的氢原子静电势为正所以偏红色,与苯环骨架碳相连的氢原子偏白色。T313与TEA相比,颜色偏蓝,这主要是由于BF3基团的存在导致其部分位置的静电势偏负,而DPG、DPTU和DOTG三者静电势分布则相差不大。
图1
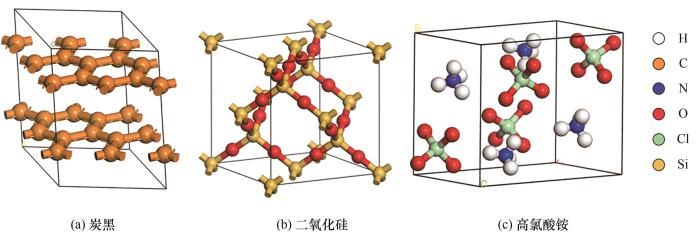
图1 纳米填料的原始晶胞
Fig.1 Primordial cells of nanofillers
图2
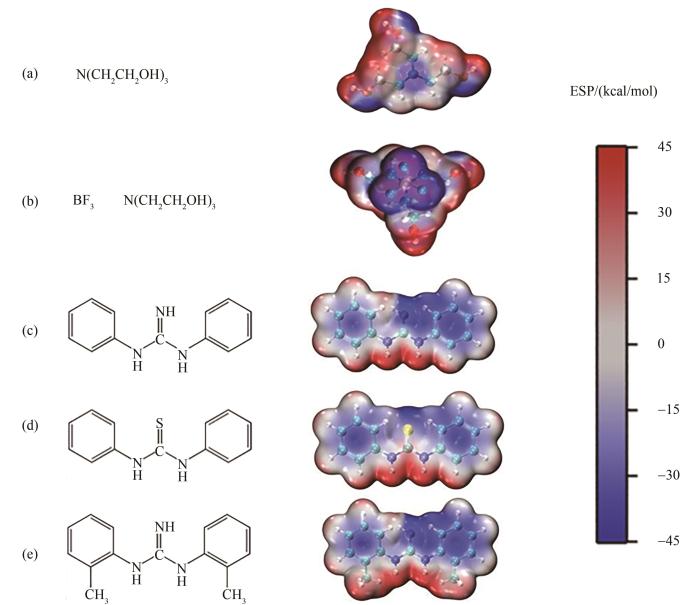
图2 五种键合剂分子的分子结构与静电势分布: (a) TEA; (b) T313; (c) DPG; (d) DPTU; (e) DOTG
(1 cal=4.184 J)
Fig.2 Molecular structure and electrostatic potential distribution of five bond agents: (a) TEA; (b) T313; (c) DPG; (d) DPTU; (e) DOTG
固体填料通过键合剂分子与聚合物主体相结合[29-30],虞振飞[31]研究了此种吸附模型。由于固体填料与聚合物之间填充了键合剂分子,固体填料与聚合物两者之间的相互作用能可忽略不计。聚合物中含有较多极性原子且属于多官能度体系,容易与键合剂分子形成吸附能更强的氢键[32]。聚合物与键合剂的吸附能远大于纳米填料与键合剂之间的吸附能,因此整个体系相容性的限制因素是键合剂与固体填料的结合能力[31]。只有遴选出一种与固体填料吸附能大的键合剂,才能提升高分子复合材料的力学性能以及较大程度防止“脱湿”现象的发生[33]。本文的计算和建模过程如下:首先对键合剂分子进行优化,然后对基底进行无定形优化,最后只将基底的第一层放开,固定下面的几层,把键合剂分子平行放置于基底上方,吸附初始距离为3.2~3.6 Å,吸附体系在计算中不断优化最终得到最优结果。DOTG、DPG和DPTU三种分子结构相似,与基底的吸附模型相差不多,因此仅放置炭黑和二氧化硅与一种键合剂的吸附模型图。TEA与T313的分子结构类似,因此放置T313与高氯酸铵的吸附模型图。图3表示DOTG在炭黑和二氧化硅上的吸附模型、T313在高氯酸铵上的吸附模型。
图3
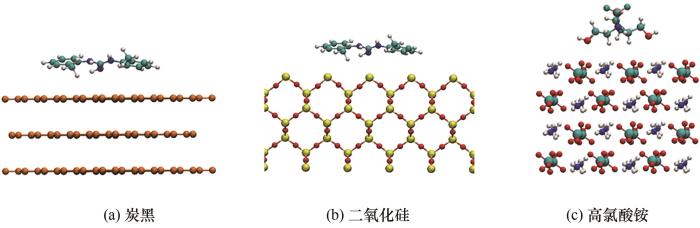
图3 键合剂在不同纳米填料表面的吸附模型
Fig.3 Adsorption model of bond agent on surface of different nano-fillers
吸附能的计算公式为
式中,
2 结果与讨论
2.1 键合剂在纳米填料表面的吸附
2.1.1 键合剂分子在不同层数高氯酸铵表面的吸附能
纳米高氯酸铵填料表面与TEA和T313的吸附能见图4,数值越大,吸附能越大。TEA分子与单层高氯酸铵的吸附能为-0.72 eV;当高氯酸铵增加到两层时,吸附能变为-0.84 eV,吸附能数值增加了0.12 eV;进一步增加高氯酸铵基底层数,吸附能不再发生明显改变。而T313分子与单层高氯酸铵的吸附能为-1.18 eV,当高氯酸铵的厚度增加到三层时,吸附能达到稳定值,此时的吸附能为-1.37 eV。对比发现,高氯酸铵对T313分子有更强的吸附能,吸附能数值比TEA大0.53 eV。造成这一现象的主要原因是T313分子中含有电负性较大的氟原子,电子云密度较高,更易吸附在高氯酸铵表面。
图4
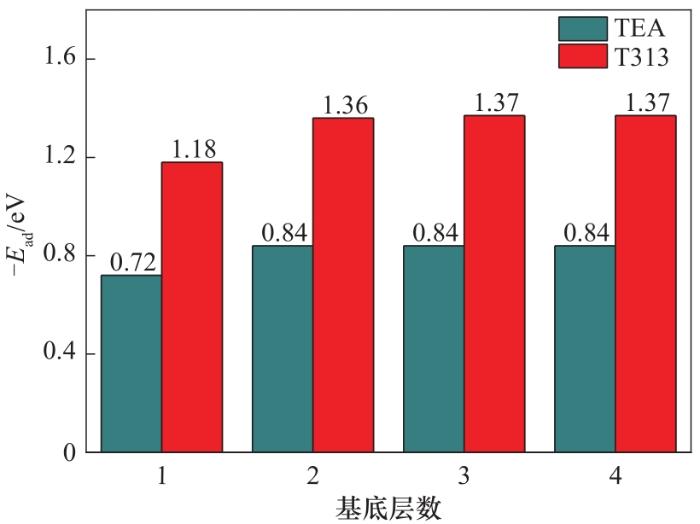
图4 键合剂在不同层数的高氯酸铵上的吸附能
Fig.4 Adsorption energy of bond agents on different layers of ammonium perchlorate
2.1.2 键合剂分子在不同层数炭黑表面的吸附能
DPG、DOTG和DPTU在炭黑表面的吸附能见图5。不同厚度的炭黑展现不同的吸附能。其中,DOTG分子在单层炭黑表面的吸附能为-1.27 eV,在双层炭黑表面的吸附能为-1.29 eV,当炭黑的层数增加到三层时,吸附能基本保持稳定。三种键合剂分子与不同层数炭黑基底之间的吸附能有着类似的变化趋势,即两层炭黑比一层炭黑的吸附能大,当炭黑的层数增加到第三层时,吸附能不再发生变化。对比发现,炭黑与DOTG分子的吸附能最大,部分原因是DOTG有两个甲基。甲基为供电子基团,而炭黑与有机分子之间主要是范德华力,因此吸附能较大。
图5
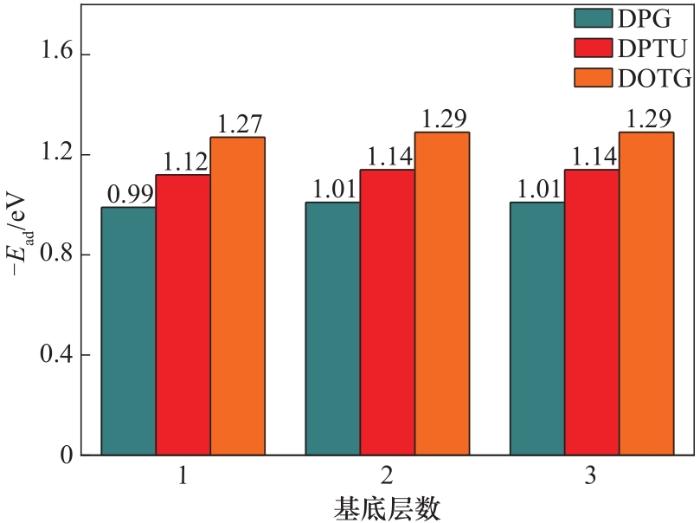
图5 键合剂在不同层数的炭黑上的吸附能
Fig.5 Adsorption of bond agents on different layers of carbon black
2.1.3 键合剂分子在不同层数二氧化硅表面的吸附能
通过计算高氯酸铵和炭黑体系发现,当基底增加到一定层数时,吸附能便不会变化,在本次计算中,发现了同样的规律。当二氧化硅达到两层后,吸附能便不再变化,见图6,二氧化硅对DOTG分子的吸附能最大为-0.94 eV,对DPG分子的吸附能最小,其稳定值为-0.87 eV。
图6
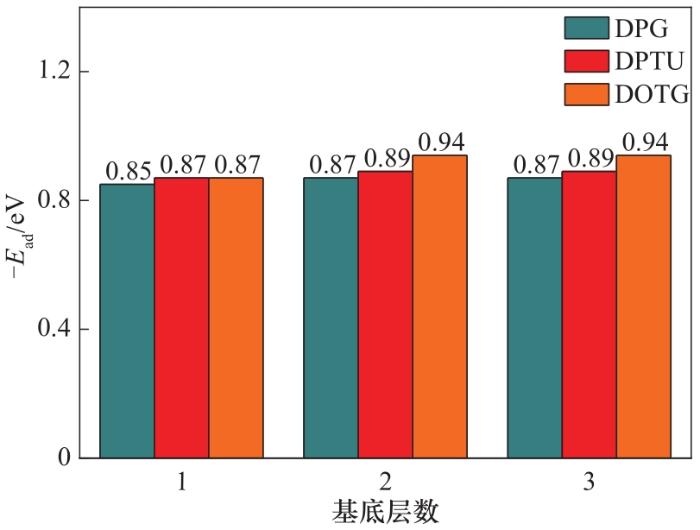
图6 键合剂在不同层数的二氧化硅上的吸附能
Fig.6 Adsorption of bond agents on different layers of SiO2
2.2 炭黑表面缺陷对键合剂吸附的影响
轮胎的加工需要诸多工艺流程。橡胶需要密炼、开链、硫化、压模等工序才能制造出成品。硫化是轮胎生产过程中必不可缺的工艺,会使橡胶获得更好的加工性能及力学性能。硫化过程中的温度会达到140℃,容易在炭黑表面局部氧化[34]或者产生不同的缺陷位[14,35-36]。基底材料的缺陷会改变对分子的吸附特性。研究了三种缺陷(单空位缺陷、双空位缺陷、Stone-Wales缺陷)位点处对吸附能的影响,三种缺陷模型见图7,为方便观察,缺陷周边用绿色表示。
图7
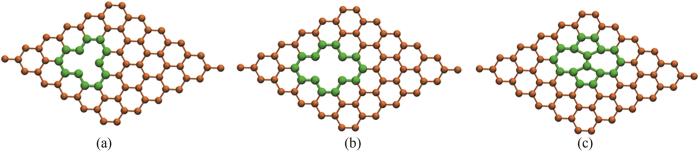
图7 炭黑表面缺陷模型: (a) 单空位缺陷;(b) 双空位缺陷;(c) Stone-Wales缺陷
Fig.7 Carbon black surface defect model: (a) single vacancy defect; (b) divacancy defect; (c) Stone-Wales defect
三种键合剂分子在不同缺陷类型炭黑表面的吸附能计算结果见表1。研究发现,单空位缺陷、双空位缺陷炭黑都要比无缺陷的炭黑的吸附能小,但并不明显,而Stone-Wales缺陷显著增加吸附能。这一现象的部分原因可以归结为Stone-Wales缺陷改变碳-碳原子的键长,从而引起电荷调制[37-39],使吸附能增加。
表1 有机分子与不同缺陷类型炭黑的吸附能
Table 1
| 缺陷类型 | 吸附能/eV | ||
|---|---|---|---|
| DPG | DPTU | DOTG | |
| 无缺陷 | -0.99 | -1.12 | -1.28 |
| 单空位 | -0.99 | -1.12 | -1.27 |
| 双空位 | -0.97 | -1.09 | -1.26 |
| Stone-Wales | -1.01 | -1.14 | -1.30 |
新窗口打开| 下载CSV
进一步以DPTU为例分析不同炭黑表面类型与键合剂分子的相互作用。图8所示为DPTU分子与炭黑表面的弱相互作用,采用Multiwfn和VMD软件开展计算并将弱相互作用可视化。图中可以清楚地看到,具有单空位和双空位缺陷的炭黑与键合剂分子的弱相互作用几乎相同。而Stone-Wales缺陷位点的炭黑与键合剂分子的弱相互作用较大,这与前述计算结果相符。弱相互作用可视化后大部分为绿色,即属于范德华力,这与前人[17]的研究报道相符。
图8
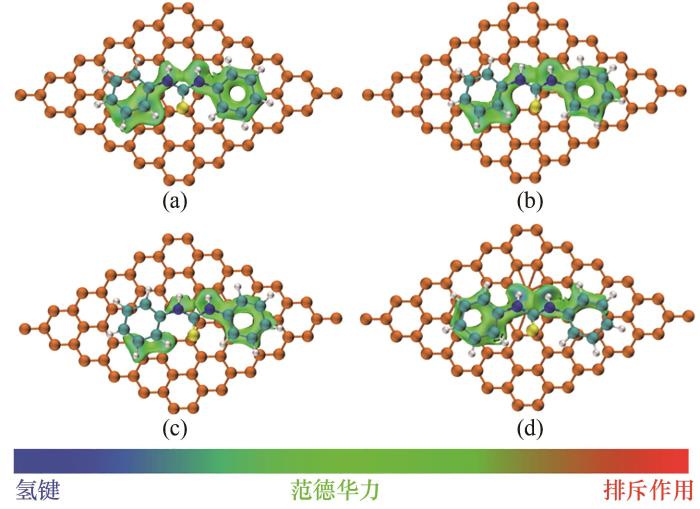
图8 DPTU在不同缺陷类型炭黑上的吸附: (a)无缺陷;(b)单空位缺陷;(c)双空位缺陷;(d)Stone-Wales缺陷
Fig.8 Adsorption of DPTU on carbon black with different defect types: (a) normal;(b) single vacancy defect;(c) divacancy defect;(d) Stone-Wales defect
2.3 SiO2表面改性对键合剂吸附的影响
二氧化硅由于含有氧元素,容易氧化在表面形成羟基[40-42]。研究了二氧化硅表面形成单生羟基对键合剂吸附的影响。计算发现,含有羟基的基底材料与键合剂分子的吸附能力显著增强。SiO2表面接枝羟基的模型见图9。SiO2表面改性前后的吸附结果对比见图10,其中SiO2@OH表示表面有羟基。键合剂分子在羟基改性表面的吸附能普遍增大,这是由于羟基容易与有机分子的极性原子形成氢键,增强了吸附作用。
图9
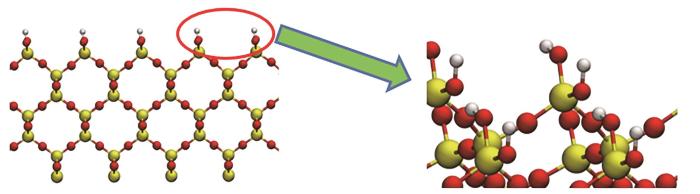
图9 二氧化硅表面改性模型
Fig.9 Surface modification model of silica
图10
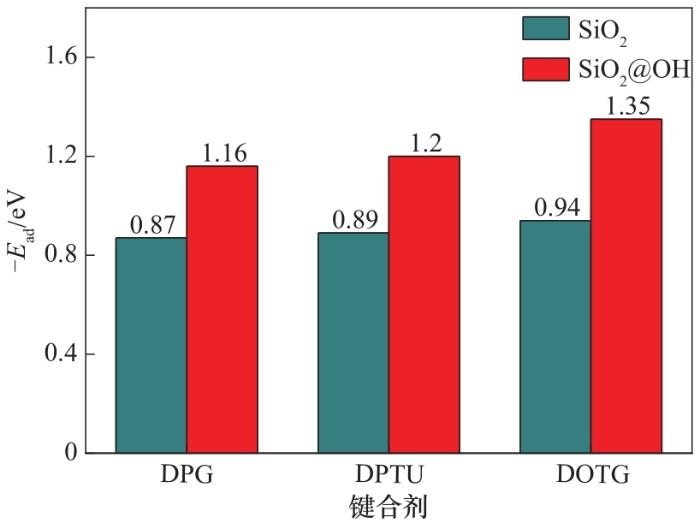
图10 三种键合剂分子在SiO2、SiO2@OH上的吸附能
Fig.10 Adsorption energy of three molecules on SiO2 and SiO2@OH
对DPG-SiO2体系、DPG-SiO2@OH体系做弱相互作用分析(图11),图中的红色圆圈部分表示氮原子与填料表面的相互作用,对比发现改性后的二氧化硅与键合剂中N原子的相互作用图会有蓝色部分,而无羟基的部分只有绿色,即改性后的二氧化硅表面的H原子与键合剂的N原子形成的氢键。由于氢键的形成对角度、距离都有严格的要求,所以图中仅有一个氢键。
图11
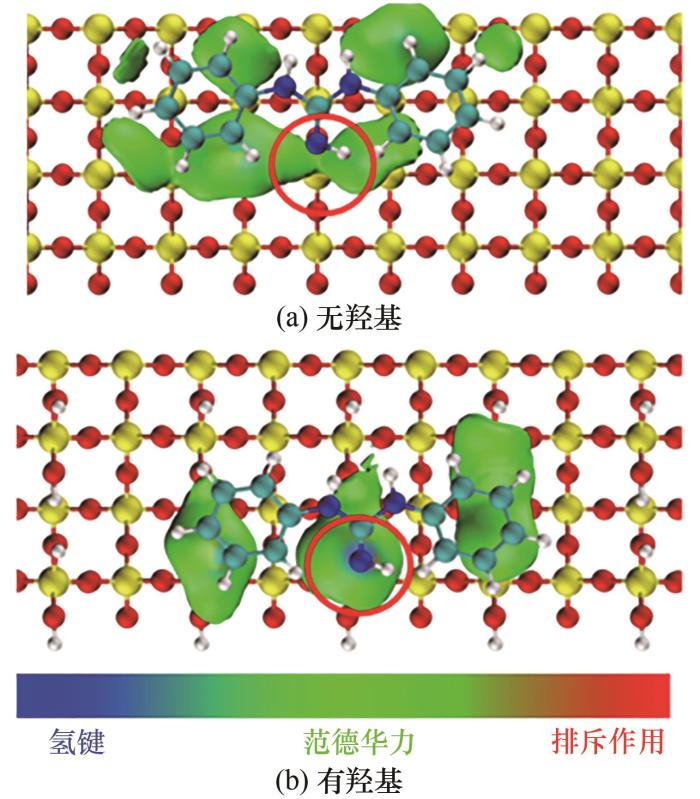
图11 DPG-SiO2体系、DPG-SiO2@OH体系的弱相互作用
Fig.11 Weak interaction of DPG-SiO2 system and DPG-SiO2@OH system
2.4 吸附能结果比较
三种键合剂分子与双层炭黑、双层SiO2、双层SiO2@OH的吸附能结果见表2。三种键合剂在双层SiO2表面的吸附能最低,为-0.87~-0.94 eV;双层炭黑基底次之,吸附能为-1.01~-1.29 eV;而在双层SiO2@OH表面的吸附能最高,为-1.16~-1.35 eV。炭黑主要依靠范德华力吸附键合剂,而改性后的二氧化硅表面有羟基,对含有极性原子的键合剂有较强的吸附特性,将二氧化硅表面的羟基去掉后,计算结果表明它对键合剂的吸附能要比炭黑小。
表2 有机分子与不同基底的吸附能
Table 2
| 基底类型 | 吸附能/eV | ||
|---|---|---|---|
| DPG | DPTU | DOTG | |
| 双层炭黑 | -1.01 | -1.14 | -1.29 |
| 双层SiO2 | -0.87 | -0.89 | -0.94 |
| 双层SiO2@OH | -1.16 | -1.20 | -1.35 |
新窗口打开| 下载CSV
3 结论
键合剂分子与固体填料的相互作用能是影响高分子复合材料的重要因素,从而是筛选键合剂或固体填料的重要依据。本文采用第一性原理计算研究了键合剂分子与高氯酸铵、炭黑和二氧化硅的吸附能。研究发现:(1)吸附能都随固体基底层数的增加而增大,但三层后就基本趋近于一个稳定值;(2)炭黑单空位或双空位缺陷会轻微降低键合剂的吸附能,但Stone-Wales缺陷会明显增加吸附能;(3)在含能高分子体系中,T313与高氯酸铵的吸附能最大,更容易填充在聚合物与填料之间强化高分子主体与氧化剂固体填料的相互作用;(4)在轮胎体系中羟基改性后的二氧化硅表面易与键合剂形成氢键,从而强化吸附能。

- 刚刚!2026年中科院分区,公布!本次看点:中科院分区变更为新锐分区;不再单独发布预警期刊;37种期刊“under review”~
- 这些重要报纸理论版都支持邮箱投稿!回复极快!
- GB/T 7714-2025与GB/T 7714-2015相比,变更了哪些,对期刊参考文献格式有什么影响?
- 别被这个老掉牙的报纸理论版投稿邮箱误导了!最新核实91个报纸理论版投稿邮箱通道,一次集齐
- 喜报!《中国博物馆》入选CSSCI扩展版来源期刊(最新CSSCI南大核心期刊目录2025-2026版)!新入选!
- 2025年中科院分区表已公布!Scientific Reports降至三区
- 国内核心期刊分级情况概览及说明!本篇适用人群:需要发南核、北核、CSCD、科核、AMI、SCD、RCCSE期刊的学者
- CSSCI官方早就公布了最新南核目录,有心的人已经拿到并且投入使用!附南核目录新增期刊!
- 北大核心期刊目录换届,我们应该熟知的10个知识点。
- 注意,最新期刊论文格式标准已发布,论文写作规则发生重大变化!文字版GB/T 7713.2—2022 学术论文编写规则

