 0373-5939925
0373-5939925 2851259250@qq.com
2851259250@qq.com

基于分子反应动力学模拟的六甲基二硅氧烷热解机理研究
作为有高比表面积、强表面活性以及优良光学性能的纳米材料,二氧化硅纳米颗粒在生物医学、催化、半导体、光学等领域都有着广泛的应用[1-3]。火焰气相合成是制备二氧化硅纳米颗粒的最重要技术之一。有机硅六甲基二硅氧烷(HMDSO)因为沸点低、稳定性好以及燃烧无污染等特点,成为制备SiO2纳米颗粒的理想前体之一。然而火焰合成过程非常复杂,通常包含热解和氧化过程,且反应发生在极短的时间内,燃烧温度、停留时间、火焰成分、前体浓度等条件变化都会对关键产物(例如SiO2以及CH3基团)的形成造成影响,使合成的SiO2纳米颗粒的结构尺寸及表面特性发生改变[4-5]。了解六甲基二硅氧烷的热解过程和机理,一方面便于理解不同反应条件对热解产物的影响,从而制备出所需要的SiO2颗粒;另一方面有利于建立六甲基二硅氧烷的反应动力学模型并为开展数值模拟奠定基础。
目前国内外学者针对HMDSO的热解反应过程已经开展了理论和实验研究。Chernyshev等[6]在650~720℃的流动反应器内进行了HMDSO热解实验,通过对产物的检测发现HMDSO热解会产生二甲基硅酮等物质,并根据检测到的产物对HMDSO热解过程进行分析,提出了三条可能的HMDSO初始热解反应路径。Alexander等[7]则利用等离子体反应器进行了HMDSO热解实验,发现产物中存在H2、CH3、CH4等小分子产物以及一些小的含硅碎片,例如(CH3)2SiH。Chrystie等[8]研究了低压贫氧惰性气氛下的HMDSO燃烧,提出了HMDSO在H2/O2/Ar火焰中热分解的10步反应机理,进一步加深了人们对HMDSO热解过程的理解。Almond等[9]通过量子化学计算,研究分析了HMDSO初始热解过程中的反应路径,揭示了HMDSO初始热解过程中最有可能发生的反应路径为Si—C键解离导致的甲基脱离。当前实验手段只能针对少数可检测的物质(例如碳氢化合物和SiO)从宏观层面开展反应路径分析和机理研究[6-8],而很多重要热解产物目前的实验手段难以测量,所以需要开展理论计算,而量子化学计算虽然精度高,但其过高的计算成本限制了模拟规模和时间[10],且量子化学计算需要对反应路径有先验认知[11]。所以为了充分理解HMDSO的热解过程及其机理,需要采用更为合适的方法开展进一步深入的研究。
分子动力学模拟(MD)相比于量子化学计算成本更低,能够模拟更大规模的分子体系。随着近些年来ReaxFF反应力场的不断发展,其被用于研究更为复杂的反应体系的成键、断键以及电荷转移过程。ReaxFF力场的开发使用了研究体系量子化学计算数据组成的训练集,所以能以接近量子化学计算的精度进行大规模的体系模拟,且ReaxFF MD方法无须对反应路径有先验认知,可以模拟体系内分子间相互反应的过程[12-13]。因此,基于ReaxFF反应力场的分子动力学模拟方法能够以较低的计算成本对复杂反应动力学过程进行研究,例如它可以用来研究物质的热解和燃烧反应过程及机理[14-28]。目前,国内外研究人员已经开发了许多适用于C/H/O/Si体系的ReaxFF反应力场,并成功应用于不同的研究领域[29-32],但每个力场都有着自己的使用范围,已报道的力场是否适用于有机硅HMDSO热解反应体系还有待进一步研究。本文采用ReaxFF MD方法从分子角度进行大规模体系的化学反应动力学研究以揭示HMDSO的热解反应过程,首先从已报道的文献中挑选了多个针对C/H/O/Si体系的反应力场,分析该体系下不同力场的适用性,然后选择最优力场开展不同温度和压力下的HMDSO多分子体系热解模拟研究,并结合HMDSO热解气相色谱实验的产物分析,从微观与宏观两个层面深入剖析HMDSO的热解过程和机理。
1 研究方法
1.1 模拟方法
ReaxFF反应力场首先由van Duin等[11-12]基于键级理论开发而来,其采用从原子间距离得到的键级来描述当前时刻原子的连接状态,并以键级为基础计算键角、二面角、库仑力、范德华力等原子间相互作用,从而来预测研究对象的结构和性质。随着反应进行,键级不断地进行迭代更新,让ReaxFF反应力场可以正确描述复杂体系中的化学反应过程,而且计算成本相对低廉。
目前已公布的ReaxFF力场中有不少适用于C/H/O/Si体系,但即使研究体系元素组成相同,通过不同量子化学计算数据优化得到的力场参数之间也会存在着较大差异,如果未经验证贸然使用不合适的力场会产生不合理的模拟结果。本次模拟挑选了三个适用于C/H/O/Si体系且为燃烧分类的ReaxFF反应力场进行验证,其中:力场A来自Newsome等[29]对暴露于O2和H2O中的SiC表面初始氧化过程的研究,其结合了先前为C/H/O及Si/O系统开发的ReaxFF力场并使用了包含Si/Si、Si/O、Si/H、Si/C的量子力学(QM)训练集对力场进行扩展优化;力场B来自Kulkarni等[30]对O2与SiO2表面相互作用的研究,在描述SiO2-H2O界面ReaxFFSiO力场的基础上进行了扩展优化;力场C来自Soria等[31]对硅表面烷基层分解的研究,结合了先前为Si/H、C/H以及Si/C/H/O开发的ReaxFF力场,使用了包含Si/H、Si/C、C/C、C/H的量子力学训练集对力场进行扩展优化。在接下来的模拟计算中,将以A~C来代指上述对应的力场。
在模拟前,首先对HMDSO分子结构进行优化,其几何构型如图1所示。然后,将20个优化后的HMDSO分子随机分布于模拟盒中,构建的体系初始构型如图2所示,通过LAMMPS软件对体系进行能量最小化处理,然后在298 K、1 atm(1 atm=101325 Pa)条件下使用NPT系综对体系进行30 ps的压缩与弛豫。
图1
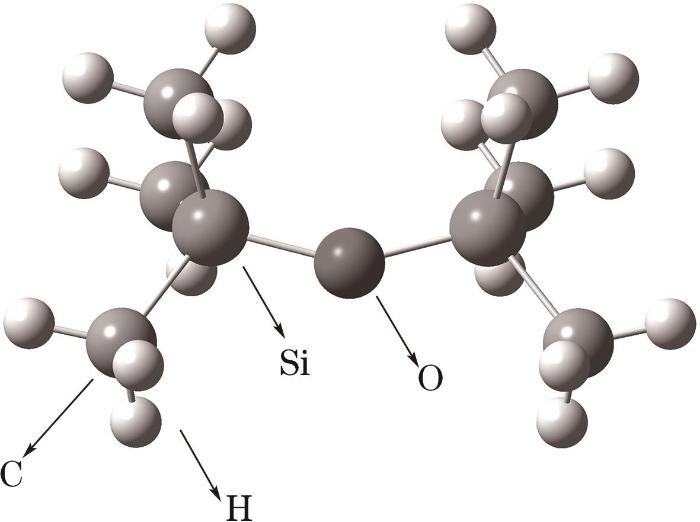
图1 HMDSO分子几何构型
Fig.1 Geometry of HMDSO molecule
图2
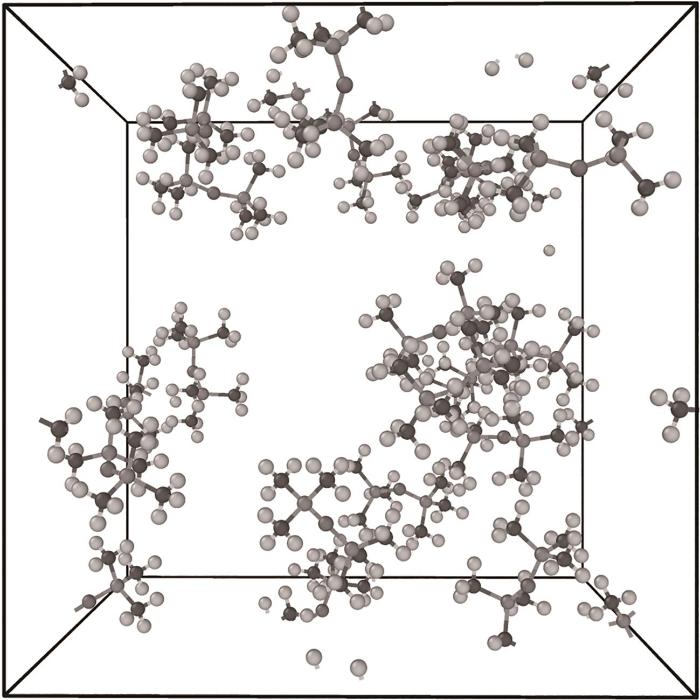
图2 HMDSO分子热解模拟体系的初始构型
Fig.2 The initial structure of HMDSO pyrolysis simulation system
为了确定在不同ReaxFF力场下HMDSO分子模型的热解温度,在LAMMPS软件中使用ReaxFF MD,针对上述三种反应力场A~C开展300~2500 K、持续500 ps的匀速升温模拟。在合适的热解温度下使用各力场对HMDSO单分子以及20个分子的体系进行500 ps的等温热解模拟,以研究不同力场条件下HMDSO热解模拟反应路径。整个模拟过程采用NVT系综,时间步长设置为0.25 fs,采用Berendsen控温,温度阻尼系数为25 fs,每800步进行一次输出,键级截止值设置为0.3 Å(1 Å=0.1 nm),通过基于原子间距离得到的键级进行产物分析。为了防止等温模拟初期由于温度的剧烈变化导致的体系波动,在模拟开始前为体系内原子设置了目标温度下基于高斯分布的初始随机速度,并挑选了三组目标温度下的等温热解模拟进行初始阶段温度演化分析,如图3所示。通过模拟前25 ps温度变化可以发现,赋予原子目标温度下的随机速度并未使温度曲线出现剧烈振荡,体系温度保持在目标温度附近波动。
图3
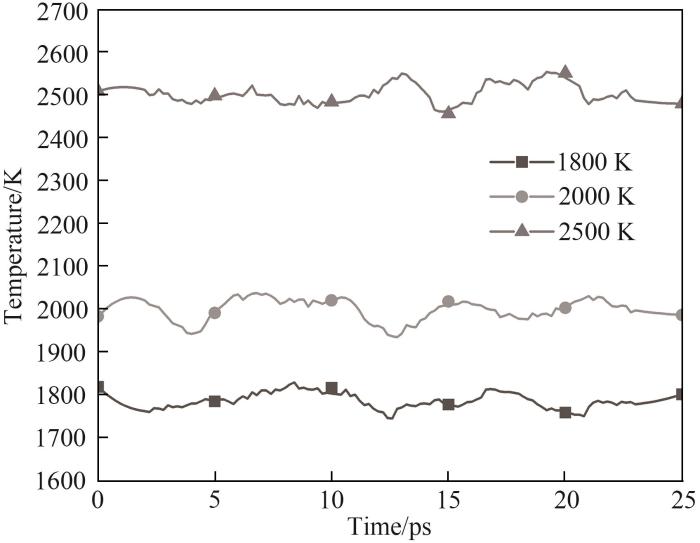
图3 不同目标温度下模拟初始阶段的温度演化
Fig.3 Temperature evolutions in the initial stage of simulations at the target temperatures of 1800 K, 2000 K and 2500 K
1.2 实验方法
向真空管式炉中通入氩气以排出石英管内其他气体。随后采用60℃恒温水浴加热HMDSO溶剂,向其中通入氩气形成载气输送到管式炉内,在700、800和900℃温度下进行三组热解实验,热解产物通过收集泵泵入采样袋进行采样。使用气相色谱仪火焰离子化检测器(FID)分析热解产物,采用外标法校准标样气体的种类及含量,根据出峰时间确定产物种类,与标样气体的峰面积之比确定产物含量。
2 结果与讨论
2.1 不同力场下的HMDSO热解升温模拟
为了了解不同ReaxFF反应力场对HMDSO热解模拟温度的影响,统计了不同力场升温模拟过程中体系HMDSO分子数随时间的变化,如图4所示。
图4
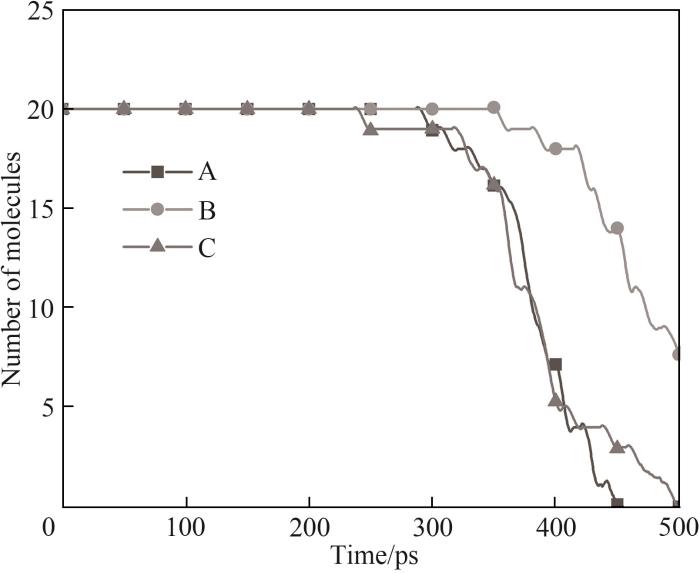
图4 不同力场在升温模拟过程中体系HMDSO分子数随时间的变化
Fig.4 Evolution of HMDSO molecular number during the heating simulation with different force fields
图4中可以看到力场A在模拟进行到300 ps左右时观察到了HMDSO分子数下降,由此可以判断此时体系发生了热解反应,而力场B与力场C热解分别发生在350 ps和250 ps左右,判断出使用力场A、B、C所对应的HMDSO初始热解温度分别在1500~1600 K之间、1800~1900 K之间以及1400~1500 K之间。图5展示了不同力场条件下升温模拟第40 ps时体系情况,此时HMDSO尚未发生热解反应,通过观察轨迹发现力场A和力场B中HMDSO分子在体系内较为分散,而力场C条件下HMDSO分子则聚拢成一团,这说明力场C的分子间作用效果强于力场A和力场B,更倾向于将分子吸引到一起。
图5
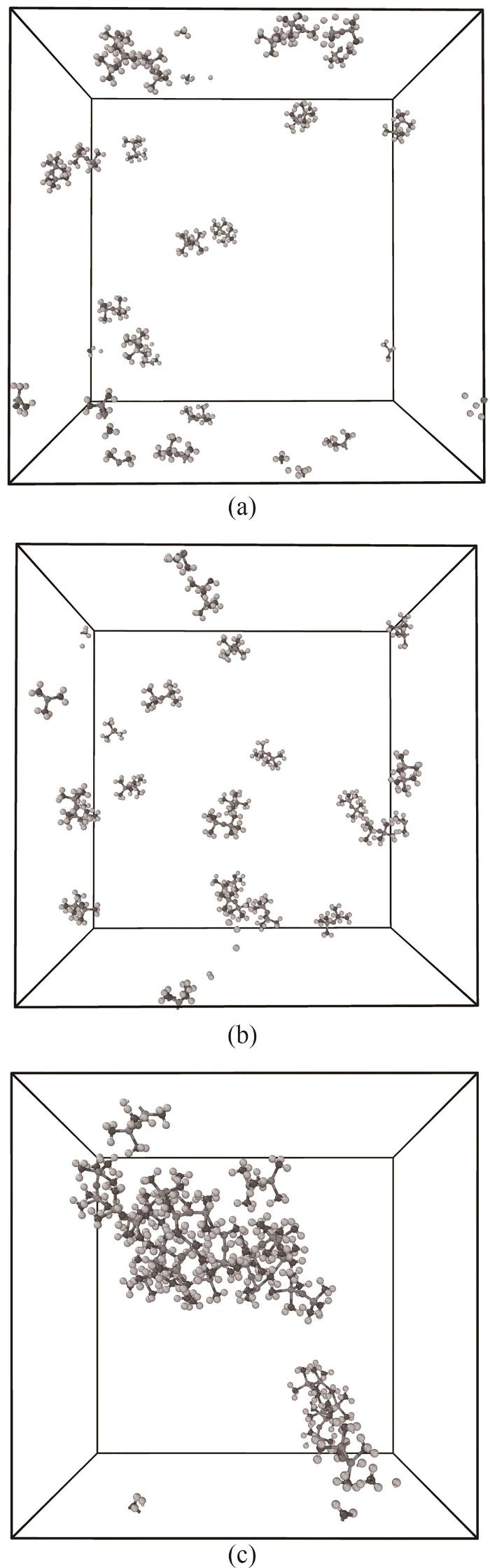
图5 模拟时间为40 ps时力场A (a)、力场B (b)及力场C (c)的体系分子分布
Fig.5 Molecular structures for force field A (a), force field B (b) and force field C (c) at 40 ps
2.2 不同力场下的HMDSO单分子热解模拟
为了清晰观察各个力场下HMDSO热解模拟过程以揭示其热解路径,对HMDSO单分子体系进行了热解模拟,每个力场在2500 K温度、NVT系综下开展十次平行模拟。
在十次平行模拟的数据统计中各个力场得出的反应路径未出现因模拟次数导致的明显差异,图6给出了使用力场A、B、C时获得的HMDSO热解初始反应步。在2500 K下,HMDSO热解反应的第一步均为Si—C键断裂导致的CH3自由基的脱离,在HMDSO初始热解反应中Si—C的键能最低[9],CH3解离是最有可能发生的反应路径,并且CH3的解离反应会随着热解过程进行不断地发生。当CH3游离在体系内时有概率与HMDSO片段发生反应,夺去氢生成CH4或是反应生成C2烃类产物,这是体系内生成CH4的主要反应。当HMDSO上氢离子被夺取后能观察到C2H4从中直接脱离。上述反应路径与先前的实验结论一致[7]。
图6
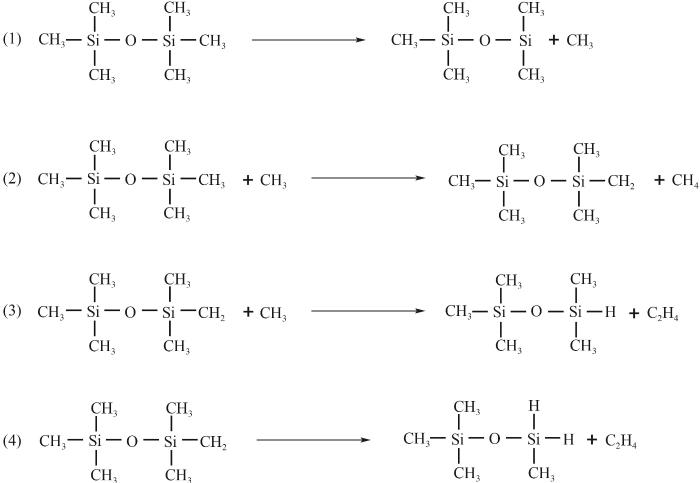
图6 HMDSO单分子的初始热解反应步
Fig.6 Primary pyrolysis pathways for decomposition of unimolecular HMDSO
力场C在热解模拟初始阶段中观察到Si—O键的断裂,HMDSO分解为二甲基硅氧烷(CH3)2SiO和四甲基硅烷(CH3)4Si,随后两个片段又迅速重组到了一起(出现于185 ps)。Si—O键断裂的解离能为504 kJ/mol,远大于Si—C键断裂的能量350 kJ/mol [9],从键能上看在HMDSO热解反应初期Si—C键断裂发生的可能性更高。由于力场C分子间更强的吸引力,能观察到键断裂形成的不稳定产物在很短时间内重组回HMDSO中,这种现象在模拟过程中反复出现,直至生成例如CH4这类稳定的热解产物。
2.3 不同力场下的HMDSO多分子热解模拟
使用不同力场对包含20个HMDSO分子的体系在NVT系综下进行2500 K的等温热解模拟。图7为不同力场条件下体系内HMDSO分子及主要产物数量随时间的变化,图8为不同力场条件下HMDSO等温热解模拟结束时体系展示。
图7
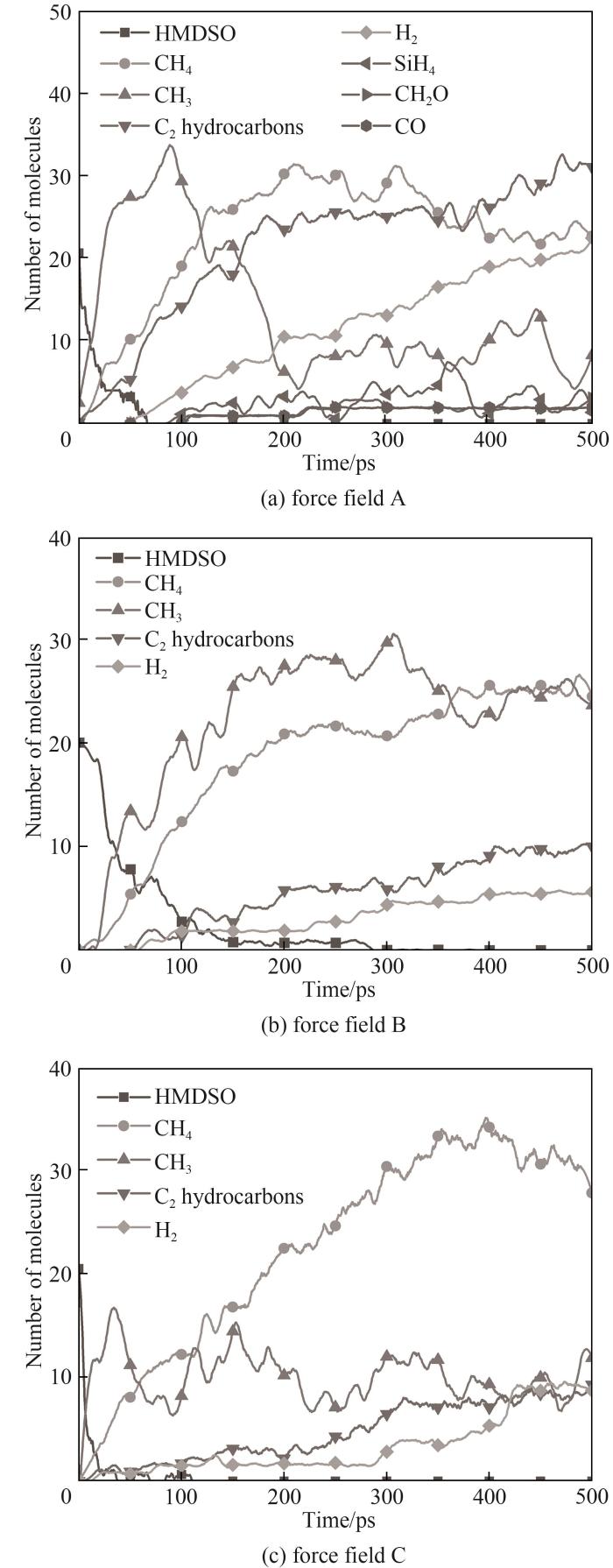
图7 不同力场条件下主要产物随时间的变化
Fig.7 Evolution of the main products during the simulations with different force fields
图8
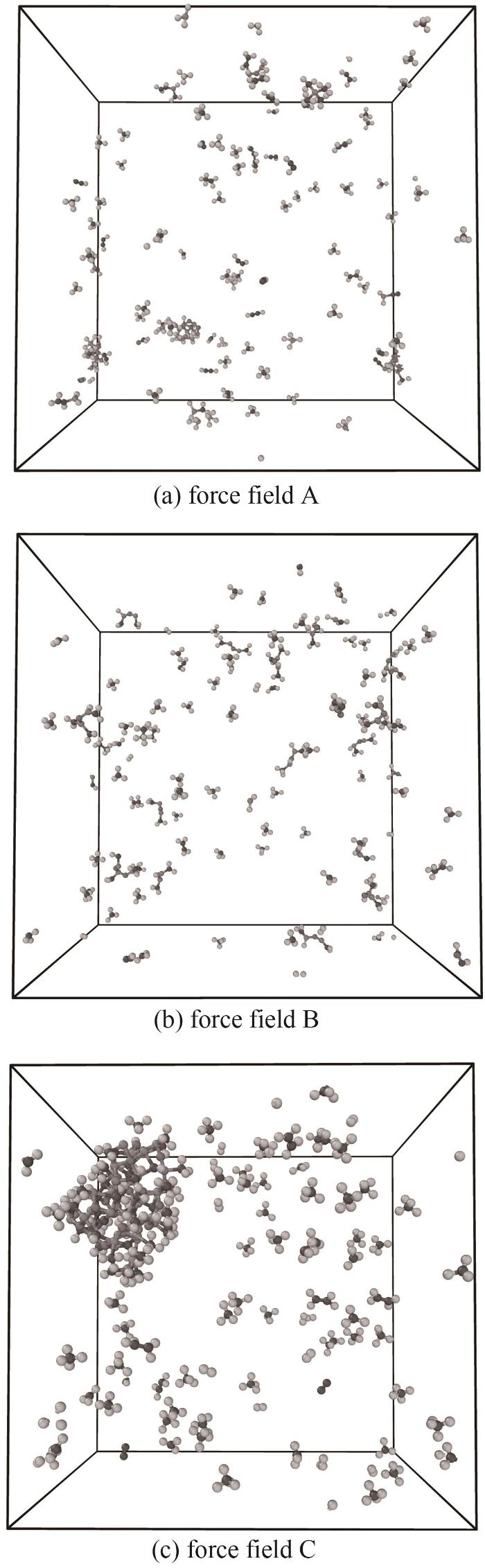
图8 不同力场条件下HMDSO模拟结束时体系分子分布
Fig.8 Molecular structures of pyrolysis products at the end of the simulation with different force fields
三个力场的主要小分子产物均为CH3、CH4、Si/C/H小分子片段及C2H2、C2H4等C2烃,与实验结果相符。力场A观察到了更丰富的小分子产物,例如SiH4及CH2O,力场B与力场C则未观察到明显的Si/H和C/H/O体系反应产物,而在实验中检测到了C/H/O体系产物[7-8]。Feroughi 等[4]在进行HMDSO低压贫氧燃烧合成SiO2颗粒以及Chrystie等[8]在低压贫氧惰性气氛下的HMDSO燃烧研究中均提及SiO组分的重要性,所以本文也对SiO产物进行了检测。只在力场A下才观察到了SiO碎片的出现(出现于148 ps),其持续时间基本不超过5ps,对轨迹进一步分析后发现,SiO产生的原因是一些中间产物上的Si—C键全部断裂导致,但SiO本身极不稳定,会在极短时间内与体系内其他活跃的分子碎片(例如CH3、Si2H4等)重组为其他物质,力场B与力场C均没有观察到这类重要反应。
截取了模拟结束时可视化体系展示图,可以发现力场C的HMDSO热解片段聚合形成了整块团簇,如图8(c)所示。进行的等温模拟末期并未进行退火,在持续高温且没有增压的情况下出现如此剧烈的成团现象并不合理,力场C表现出的极强的分子间相互作用表明其并不适合用于该体系的模拟。
综上,力场A模拟结果与先前的实验和模拟研究结果较为吻合,相较于其他现有力场更适用于HMDSO热解体系模拟。
2.4 温度对HMDSO热解反应的影响
根据升温模拟得到的初始热解温度,使用力场A分别在温度1600、1800、2000、2500 K,压力1 atm的条件下对HMDSO分子体系进行500 ps的NVT系综等温模拟,模拟参数设置与上文相同。图9展示了不同温度下体系部分主要产物随时间的变化,图10展示了不同温度下体系主要含Si产物随时间的变化。模拟产物主要为CH3、CH4、C2烃、H2、CH2O、小分子Si/H、C/Si/H含Si化合物(例如SiH4、CH4Si等)和尚未完全裂解的分子片段,在高温下还能观察到CO和C2的生成。
图9
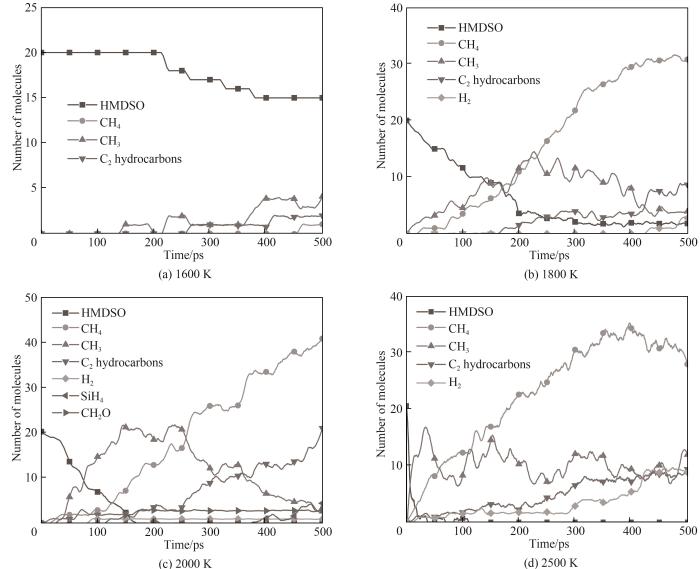
图9 不同温度下部分主要产物随时间的变化
Fig.9 Evolution of some main products during the simulations with different temperatures
图10
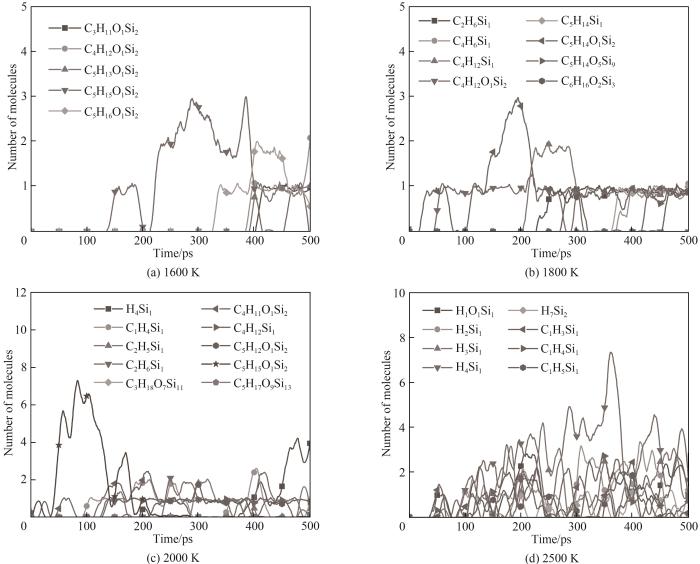
图10 不同温度下主要含Si产物随时间的变化
Fig.10 Evolution of the main Si-containing products during the simulations with different temperatures
1600 K温度下体系内主要反应为Si—C键的断裂导致的CH3自由基脱离,与上文HMDSO单分子初始热解反应结论相一致。CH3由于自身较为活跃的化学性质,在体系内运动时会与其他分子进行反应与重组,CH3可能与自由基等活跃小分子结合形成更为稳定的分子结构;也可能与HMDSO或其片段反应生成较为稳定的CH4或C2烃进而脱离;还有可能进行Si—C键的重组回存在空位的HMDSO热解片段上,这些是生成热解产物CH4及C2烃的主要反应。可以看出不同温度下体系内CH3数量变化基本呈现先上升后下降的趋势,这与HMDSO热解过程Si—C键的断裂和CH3后续的中间反应速率变化有关,温度越高其分子数变化幅度越明显,在1600 K下CH3数量缓慢上升且未出现大幅下降是因为刚达到热解温度导致的体系内反应不活跃。除了CH3还观察到了CH4从HMDSO上脱离,这是由于Si—C键断裂后CH3自由基夺取H进行重组脱离,模拟中该反应观察到的频率远不及Si—C键断裂反应生成CH3,这与量子化学计算得到的CH4脱离所需克服的能垒高于CH3的脱离结论相一致[9]。在热解反应中观察到H转移的现象(1600 K温度下出现于53 ps),例如脱去了一个甲基的(CH3)3SiOSi(CH3)2会从其他HMDSO分子或片段上夺取一个H填补在空位上形成(CH3)3SiOSi(CH3)2H。
随着热解温度升高体系内观察到了更多重要反应。高温下HMDSO发生了Si—O键的断裂,生成了Si(CH3)4以及(CH3)2SiO (1800 K温度下出现于198 ps);观察到了C2H2从HMDSO热解片段中脱离。随着热解反应的继续进行,Si—O键断裂的情况逐渐增多,并且生成的分子片段有概率进行重组聚集成大型团簇,这与Chernyshev等[6]进行的HMDSO热解反应的实验结果一致。在整个模拟过程中只观察到了Si(CH3)4的生成,体系中并没有发现Si(CH3)3,这也与HMDSO热解实验产物分析结果相一致。由此可以推断HMDSO热解中Si—O键断裂反应倾向于形成更为稳定的Si(CH3)4而非Si(CH3)3,HMDSO热解为Si(CH3)4和(CH3)2SiO的反应解离能比热解为Si(CH3)3和(CH3)3SiO更低[9]。只在较高温度下观察到了SiO的生成(2500 K温度下出现于148 ps),在整个模拟过程中多次出现又很快反应消失。当热解温度很高时体系内分子运动极为活跃使得热解分子片段间的重组反应难以发生,体系内观测不到大分子团簇,相较于其他模拟温度整体体系呈现完全碎片化的趋势,如图11所示。
图11
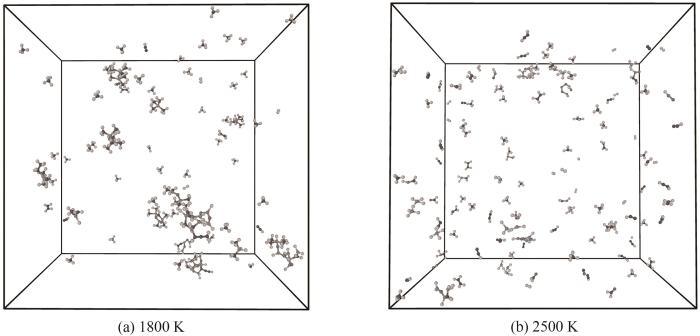
图11 1800 K与2500 K温度条件模拟体系展示
Fig.11 Molecular structures of pyrolysis products at 1800 K and 2500 K
2.5 压力对HMDSO热解反应的影响
使用NPT系综在温度298 K、压力分别设置为0.1、0.5、1、2、4 atm的条件下对体系进行30 ps的缩放与弛豫,弛豫结束后对应的模拟盒尺寸分别为119.2、84.3、70.4、62.4、49.3 Å,对应HMDSO浓度0.0032、0.0090、0.0155、0.0222、0.0449 g/cm3,在1800 K的温度下使用力场A对弛豫后的各体系进行等温模拟。不同压力下模拟体系总分子数随时间变化如图12所示,体系势能随时间变化如图13所示,而图14与图15分别对应了不同压力模拟主要产物以及主要含Si产物随时间的分布情况。
图12
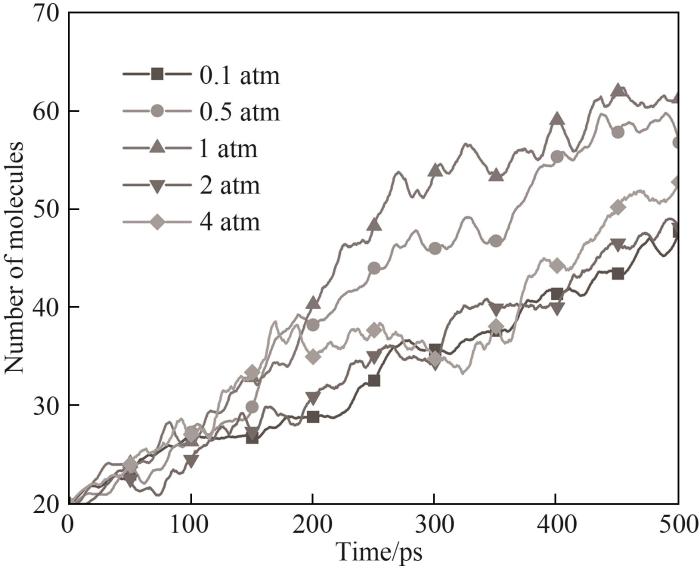
图12 不同压力下模拟体系总分子数随时间的变化
Fig.12 Evolution of the total molecular number of the system for different pressures
图13
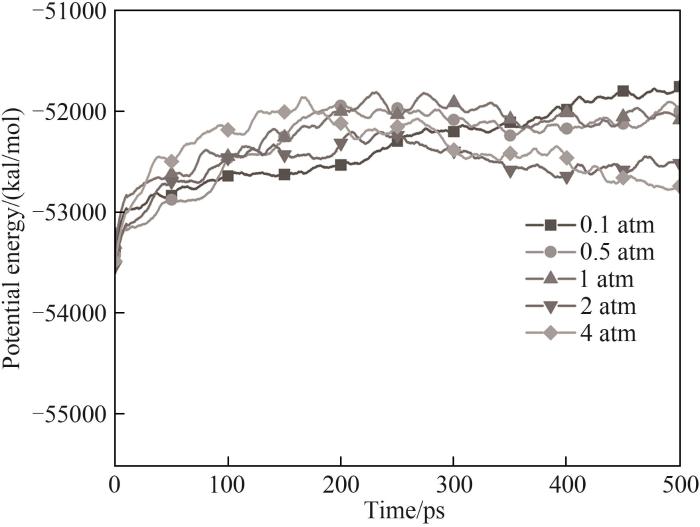
图13 不同压力下模拟体系势能随时间的变化
Fig.13 Evolution of potential energy during the whole simulation process with different pressures (1 cal=4.18 J)
图14
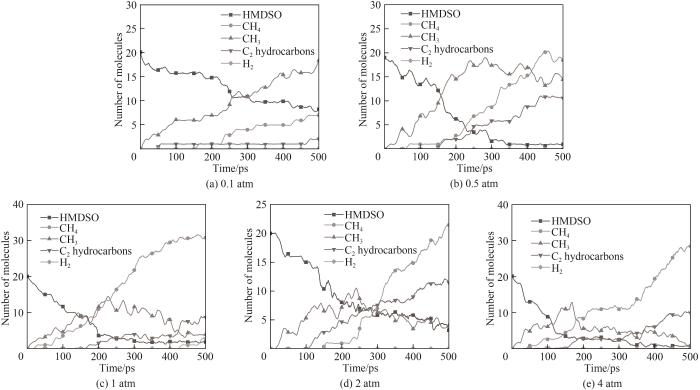
图14 不同压力下部分主要产物随时间的变化
Fig.14 Evolution of some main small molecule products for different pressures
图15
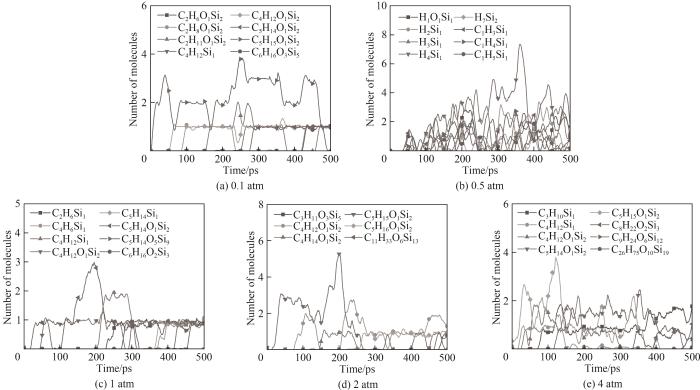
图15 不同压力下主要含Si产物随时间的变化
Fig.15 Evolution of the main Si-containing products during the simulations with different pressures
体系势能最初都经历了一段上升期,这是由于吸热反应导致的。除了0.1 atm外,其他条件下体系经过中间反应阶段后势能趋于渐近值,这意味着体系内化学反应剧烈程度不断下降,同时伴随着稳定的最终产物的生成,而0.1 atm条件下势能保持缓慢上升,在模拟结束时体系势能最高。图14中能观察到在0.1 atm条件下体系内CH3自由基数量不断上升,而其他压力下CH3都呈现先上升后减小的趋势。通过对产物和轨迹的分析发现0.1 atm下由于体系内较低的分子浓度,使得HMDSO经过初始热解阶段后分子间发生后续反应的概率变低,其体系分子总数和种类相比于高压条件更少,但有更多包括CH3在内的活跃分子在体系内运动,而在高压时CH3被快速地消耗并且有更多稳定的反应产物生成,据此可以推测在低压条件下HMDSO热解产物表面会因不完全热解接枝聚合更多的自由基。体系内团簇的形成也与压力密切相关,在0.1 atm下并未观察到明显的聚合成团现象,含Si产物基本上是HMDSO热解断裂形成的片段。随着模拟压力增加开始形成团簇,且压力越大体系分子越密集,形成的团簇尺寸也越大,图16展示的不同压力条件下模拟体系图能很直观地看出压力对于体系成团的影响。
图16

图16 不同压力条件下模拟体系分子分布
Fig.16 Molecular structures of pyrolysis products at different pressures
模拟结果表明压力对于HMDSO热解的影响主要体现在中间反应程度上。压力越大体系越拥挤,分子间发生反应的可能性就越大,不稳定的中间体会被快速消耗形成更多稳定的热解产物,体系内成团现象也会更加剧烈。
2.6 气相色谱实验
为了检测HMDSO热解产物以验证模拟结果的准确性,开展了气相色谱实验。气相色谱仪预设了包括烷烃、烯烃、乙炔等在内的部分常见且不超过五个碳的烃类,如表1所示。图17为不同温度下HMDSO热解气相色谱图。
表1 气相色谱仪预设化合物名及结构
Table 1
| Peak entry | Chemical name | Chemical structure | Peak entry | Chemical name | Chemical structure |
|---|---|---|---|---|---|
| 1 | methane | 2 | ethane | ||
| 3 | ethylene | 4 | propane | ||
| 5 | cyclopropane | 6 | propylene | ||
| 7 | iso-butane | 8 | n-butane | ||
| 9 | 1,2-propadiene | 10 | 1-butene | ||
| 11 | trans-2-butene | 12 | 2-methylpropene | ||
| 13 | iso-pentane | 14 | cis-2-butene | ||
| 15 | n-pentane | 16 | 1,3-butadiene |
新窗口打开| 下载CSV
图17
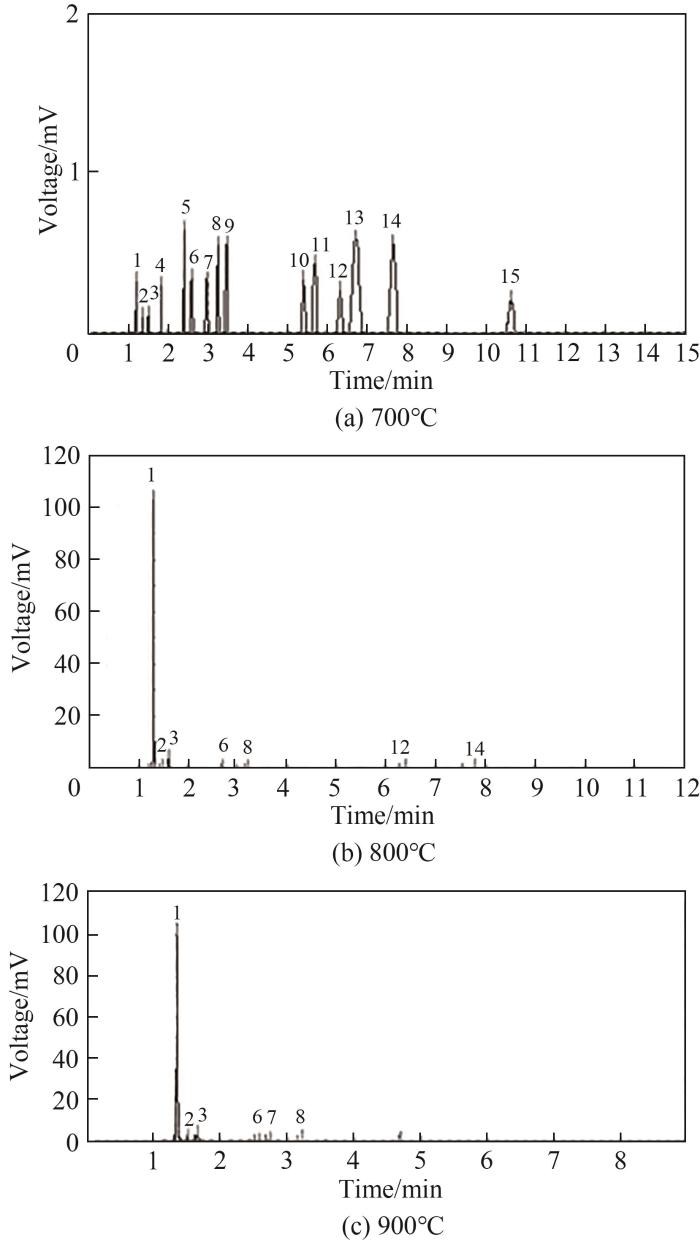
图17 温度为700℃、800℃、900℃时HMDSO热解色谱图
Fig.17 Chromatography of pyrolysis at 700℃, 800℃, 900℃
热解温度700℃时检测出15种热解产物,产物种类相比800℃与900℃时更丰富,但是烃类总含量只占产物的不到0.5%,且结构稳定的CH4、C2烃等热解最终产物与在高温下会发生裂解的高碳烃类在总产物中的含量占比相差不大,HMDSO在此温度下尚未完全热解。热解温度800℃时产物中检测出的烃类产物种类急剧减少,CH4含量提升最多成为主要烃类产物,C2H4含量小幅提升,C2H6含量变化不大,峰4~15对应的产物含量下降或未检测出,900℃时的热解产物与800℃时差别不大。总体来说随着温度升高,HMDSO热解生成的烃类产物种类明显减少,大部分为CH4和C2烃,表2给出了不同温度下ReaxFF MD模拟中主要烃类产物占比,其产物种类和趋势与实验结果基本一致,实验中统计烃类产物含量占比时用作载气的惰性气体也参与了统计,所以实验烃类产物含量占比普遍比模拟结果低。值得注意的是在2500 K模拟时CH4在产物中的含量占比相较于1800 K和2000 K时有所降低。这种现象主要由于等温热解模拟研究时并未采用升温模拟策略,使得热解温度很高时部分势垒相对较高的反应能在模拟前中期发生,且高温下产物趋于碎片化,体系总产物数量剧增,而实验过程经历了升温过程和退火过程,这会导致高温时模拟产物占比与实验产生较大差异。
表2 不同温度下ReaxFF MD模拟部分主要烃类产物占总产物分子数的比例
Table 2
| Chemical name | Percentage/% | |||
|---|---|---|---|---|
| 1600 K | 1800 K | 2000 K | 2500 K | |
| methane | 3.70 | 49.21 | 48.19 | 19.83 |
| acetylene | 0 | 3.17 | 12.05 | 10.34 |
| ethylene | 3.70 | 7.94 | 6.02 | 8.62 |
| ethane | 3.70 | 0 | 1.20 | 0.86 |
新窗口打开| 下载CSV
2.7 热解反应网络
通过观察ReaxFF MD模拟轨迹以及产物分析,结合气相色谱实验数据,给出了HMDSO热解的主要反应路径,如图18所示。从图中反应网络可以看出,HMDSO初始热解路径包括CH3的解离、CH4的解离、Si—O键断裂分解为(CH3)2SiO和Si(CH3)4,以及与其他物质结合形成更大的分子结构。在这些反应路径后面,大部分的反应是通过CH3、CH4等碳氢化合物从HMDSO上断键脱离或是与之重组实现,这些反应贯穿了整个HMDSO热解反应网络,是热解产物CH3、CH4以及C2烃的主要来源,因为这些反应的能垒相对较低,反应更容易进行。HMDSO还有可能发生Si—O键断裂,从而分解为C/H/Si分子碎片及C/H/Si/O分子碎片,裂解出的碎片处于不稳定状态,会与其他碎片结合形成大的分子,在模拟中观察到的分子团簇也是由此产成。需要注意的是,重组反应不仅会发生在产物碎片之间,HMDSO分子或更大结构的分子也会与这些碎片进行重组,这些现象也可以在图18中观察到。HMDSO热解反应的主要产物除了上述的小分子碳氢化合物、裂解出的分子碎片和形成的分子团簇外,在反应后期还会生成SiH4、SiH2等硅氢化合物以及CH4Si等碳硅氢化合物,CH2O脱离后也会以比较稳定的状态存在于反应体系内。
图18
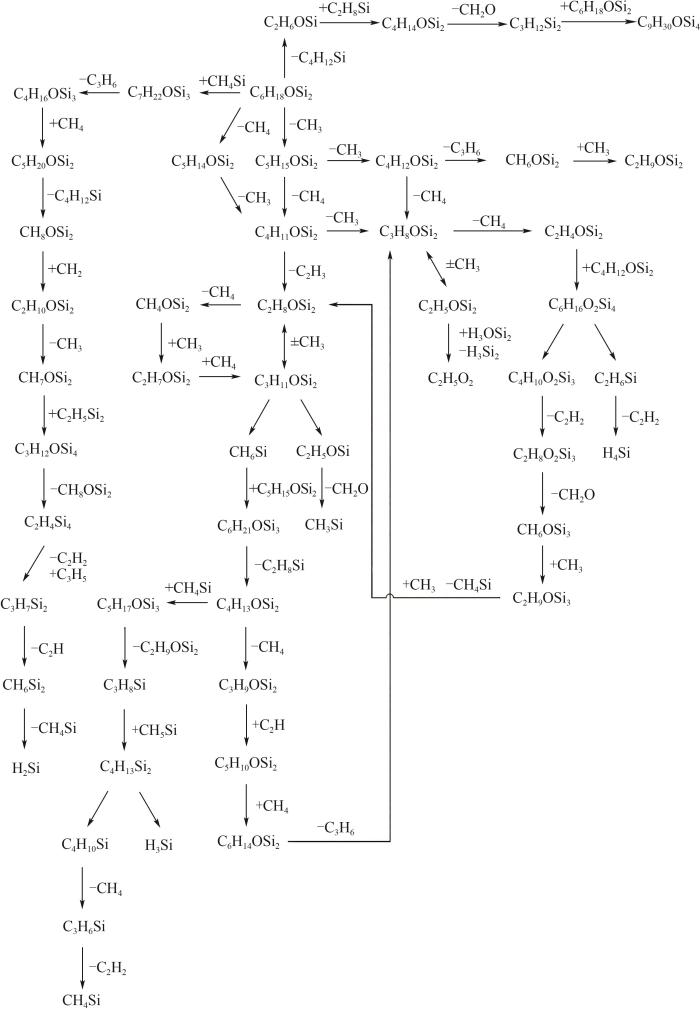
图18 HMDSO主要热解反应路径
Fig.18 Overview diagram of HMDSO pyrolysis
3 结论
本文采用ReaxFF MD方法对比了不同ReaxFF反应力场对HMDSO热解分子动力学模拟过程的影响,观察不同力场下HMDSO分子热解路径,将不同力场进行HMDSO热解模拟得到的产物分布和模拟轨迹与先前的实验与模拟结果进行对比,可以得出现有力场中Newsome等[29]针对C/H/O/Si体系开发的ReaxFF反应力场适用于HMDSO热解模拟。选定力场后进行了HMDSO热解模拟,分析了主要产物在不同温度以及不同压力条件下的形成及演变规律,并进行了HMDSO热解气相色谱实验,揭示了HMDSO的热解反应路径和机理。结果发现,温度对于HMDSO的热解反应有着重要影响,当温度达到HMDSO热解条件时,其初始热解反应为Si—C键断裂导致的CH3脱离。随着温度升高热解反应加剧,观察到H转移、CH4脱离、Si—O键断裂以及分子间重组的一系列反应,热解的主要产物为CH3、CH4、C2烃、H2、SiH4、CH2O、HMDSO初始热解片段以及一些大分子团簇。当热解温度进一步上升时,发现HMDSO会因反应过于剧烈而出现热解产物完全碎片化的现象,分子片段在此温度下不会聚集成大型分子团簇。SiO作为HMDSO高温热解下的中间产物,由于其很不稳定,在体系内生成之后会很快参与反应消失,除此之外,产物中还会出现少量CO、C2、Si/H以及C/Si/H组成的小分子物质。压力对HMDSO热解反应的影响主要体现在体系浓度变化导致的中间反应程度改变。当体系压力增加时,分子间相互碰撞的可能性变高导致中间反应更为剧烈,产物种类增多并且分子间重组成团更为明显,反之亦然。产物分析发现体系内CH3数量先上升后下降,其原因是Si—C键断裂反应势垒较低导致其在热解初期大量生成,但是CH3本身不够稳定,会与其他的分子结合而不断消耗,因此反应后期数量会逐渐下降,体系内CH3的数量变化与HMDSO初始热解反应与中间反应剧烈程度相关。

- 刚刚!2026年中科院分区,公布!本次看点:中科院分区变更为新锐分区;不再单独发布预警期刊;37种期刊“under review”~
- 这些重要报纸理论版都支持邮箱投稿!回复极快!
- GB/T 7714-2025与GB/T 7714-2015相比,变更了哪些,对期刊参考文献格式有什么影响?
- 别被这个老掉牙的报纸理论版投稿邮箱误导了!最新核实91个报纸理论版投稿邮箱通道,一次集齐
- 喜报!《中国博物馆》入选CSSCI扩展版来源期刊(最新CSSCI南大核心期刊目录2025-2026版)!新入选!
- 2025年中科院分区表已公布!Scientific Reports降至三区
- 国内核心期刊分级情况概览及说明!本篇适用人群:需要发南核、北核、CSCD、科核、AMI、SCD、RCCSE期刊的学者
- CSSCI官方早就公布了最新南核目录,有心的人已经拿到并且投入使用!附南核目录新增期刊!
- 北大核心期刊目录换届,我们应该熟知的10个知识点。
- 注意,最新期刊论文格式标准已发布,论文写作规则发生重大变化!文字版GB/T 7713.2—2022 学术论文编写规则

